Every year, over 300 000 children are born with sickle cell disease (SCD) (Modell and Darlison, 2008). In the UK, there are approximately 12 500 people suffering with a sickle cell disorder (Sickle Cell Society, 2008). Traditionally, SCD sufferers were centred around London and the West Midlands, but the demographics have changed and there is now a more widespread distribution of cases all over the UK (Lucas et al, 2008).
SCD is characterized by acute painful crises, haemolytic anaemia, multiple organ complications and a reduced life span. Platt et al's (1994) study found the average life expectancy for males with SCD is 42 years, compared with 48 years for females. Although life expectancy has increased slightly since these estimates, it is still significantly below average. SCD is most common among those people whose families originate from Africa, Saudi Arabia, India, Central America, South America and parts of the Caribbean, and it normally presents during childhood (Hoffbrand et al, 2005).
SCD is a group of genetic blood disorders aptly named due to the morphological change that takes place in the red blood cells (RBC), turning them from their normal disk shape into a sickle-like structure. SCD manifests in several forms, but the most common and widely known form of SCD is sickle cell anaemia (SCA), also known as HbSS (Peterson, 2008). Other clinically significant forms of SCD include sickle cell trait (HbAS), sickle haemoglobin C (HbSC), sickle haemoglobin D (HbSD), and sickle £-thalassaemia (HbS £-Thal). This article will focus on HbSS as it is the most common in the UK.
Sufferers of SCD make up a small minority of the total population and, combined with improvements in local NHS services, means they rarely call upon the services of ambulance trusts, but when they do they require prompt assessment and management. This article will explore the importance of early assessment and management of symptomatic patients using current guidelines and recommendations.
Background
The first published description of SCD was given by Herrick (1910). Herrick took a sample of blood from Walter Noel Clement, a dental student, who had moved to Chicago from the West Indies in 1904 to study dentistry. Clement became repeatedly ill with shortness of breath, palpitations and episodes of icterus—jaundice seen in the sclera of the eye. On looking at Clement's RBC, Herrick was surprised to see they were elongated and sickle-shaped.
Since then, and certainly in recent years, much research has been undertaken to understand the disease and management approaches. The treatment and management of SCD patients has moved from inpatient to outpatient care due to changes in treatment and prophylactic medication. Inpatient care is usually reserved for severe acute episodes (Lucas et al, 2008). All SCD patients are referred to a specialist centre for supervision of care, with local hospitals and GPs being guided by the specialist centre.
Also in the last decade, there have been several key publications from the British Committee for Standards in Haematology (2003), the Sickle Society (2008), Lucas et al (2008) and the NHS sickle cell and thalassaemia screening programme (2010). All of these publications give guidance to practitioners on how to best manage SCD patients. The guidelines also aim to provide uniformity so all patients can expect the same level of care, regardless of their geographical location or point of access.
The Joint Royal Colleges Liaison Committee (JRCALC) (2009a) have also published updated guidelines for prehospital management of SCD patients, helping practitioners to provide treatment before arrival at hospital. Although most of the guidelines are aimed at hospital and community-based management of SCD patients, there are key themes that can equally be applied to prehospital emergency care. There are also, currently in development, National Institute for Health and Clinical Excellence (NICE) guidelines for the treatment of a sickle cell crisis in hospital (NICE, 2011).
Pathophysiology
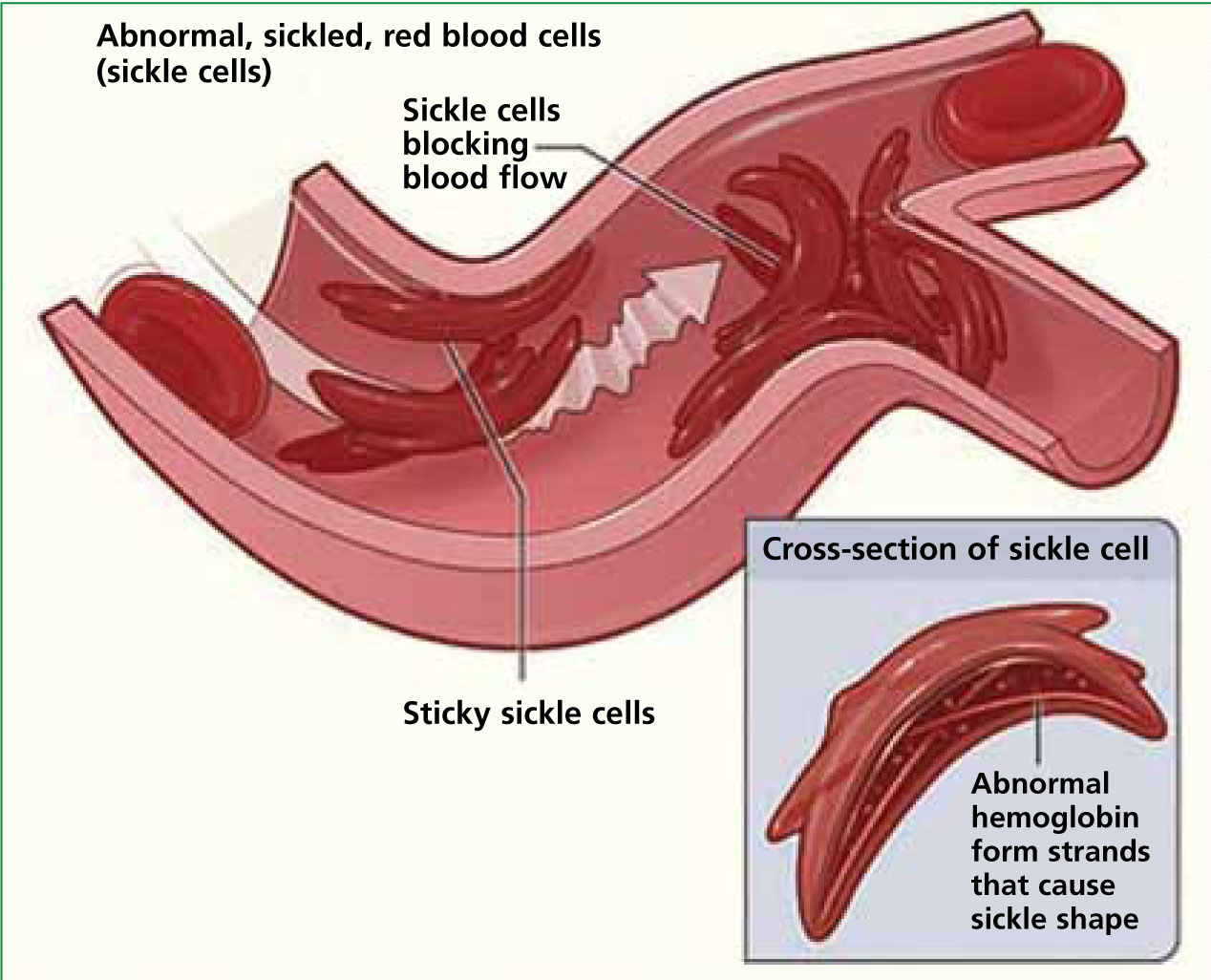
Normal RBCs, in their circular and flexible state, can travel through the circulatory system delivering oxygen unhindered. However, as a result of SCA, there is an abnormal variant of haemoglobin called HbS. This is caused by a mutation in the beta-globin chain of the haemoglobin molecule. When this abnormal haemoglobin becomes deoxygenated, it polymerizes the RBC into a sickle shape. As sickling progresses, the sickled RBCs stick together, giving rise to acute complications such as a vaso-occulsive crisis (VOC) and acute chest syndrome. RBCs can return to their normal shape with oxygenation in the lungs; but, with repeated episodes of sickling, net water and potassium loss occurs, resulting in a dehydrated and permanently damaged cell.
‘Sickle cell disease is characterized by acute painful crises, haemolytic anaemia, multiple organ complications, and a reduced life span’
The result of the damage is that cells are no longer able to return to their original shape when reoxygenated, so cells remain in a permanent sickled shape (De, 2005). The life span of a RBC is also reduced to an average of 20 days, compared with 120 days. This can lead to haemolytic anaemia (Porth and Matfin, 2010). There are many variants of abnormal haemoglobin and the collective name for these is haemoglobinopathies (De, 2008).
Genetics
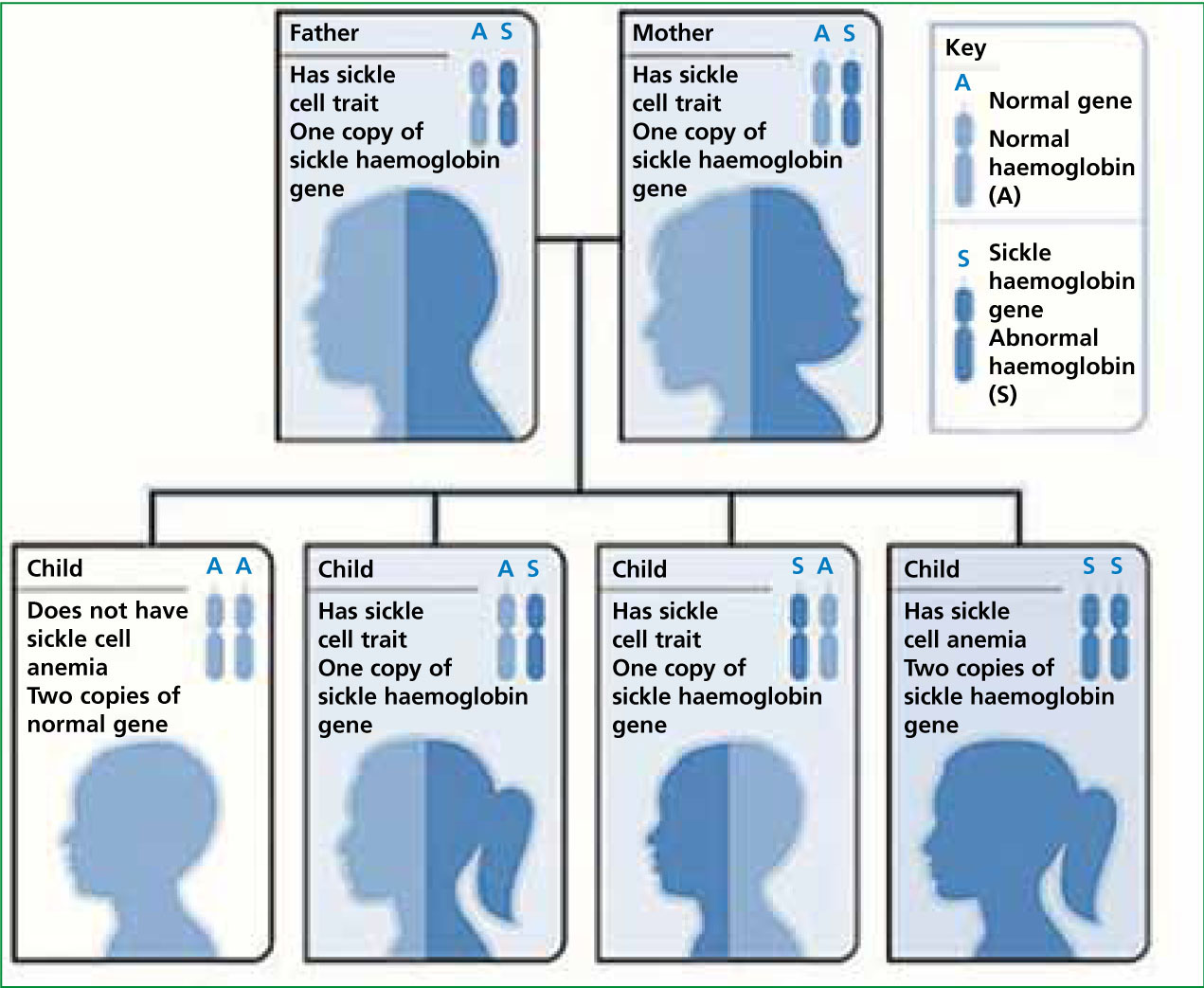
For SCA to occur, sickle cell trait (SCT) gene, HbAS, instead of a normal HbAA gene, has to be inherited from both parents. If both parents have HbAS there is a 25% chance that any children will have SCA (Figure 1). If one parent has HbAS and the other HbSS (i.e, has SCA), this will result in a 50% chance of any children having SCA. Finally, if one parent has the HbAS gene and the other has HbAA, no children will develop SCA but there is a 50% chance they will have the SCT. SCT in itself will not cause SCA and most people with SCT will be unaware that they have the condition. It is likely that any children born with SCT will remain asymptomatic throughout their lives.
Signs and symptoms
Due to the nature of the disease and its potential to affect any organ of the body, it would be unrealistic to list all the signs and symptoms that patients may present with. However, some of the most common are listed in Table 1.
| Severe pain—commonly, arms, legs, back and abdomen in adults and hands and feet in children |
| Shortness of breath, reduced Sp02, chest pain and cough indicate possible acute chest syndrome |
| Pallor/jaundice |
| Weakness |
| Dehydration |
| Prolonged painful erect penis |
| Headache/stroke |
| Fever with possible sepsis. |
Due to the multiple organ complications, it is vital THAT a full assessment is undertaken in any patient presenting with acute complications from SCD. The assessment should be guided by the patient's presenting signs and symptoms, but should follow the ABCDE stepwise approach (JRCALC, 2006), aiming to identify and correct any life-threatening sickle cell complications as early as possible.
There are several life-threatening complications of SCD which are discussed later. The clinician should be extra vigilant during patient assessment to make sure the signs are not missed. Clinicians should pay particular attention to any patients presenting with any neurological symptoms and they should receive a full neurological assessment to rule out any features of an acute stroke. Patients should also be assessed for chest pain, breathlessness, and signs of infection that may occur with acute chest syndrome.
An abdominal assessment should be undertaken in any unwell child, looking for pain, tenderness and enlargement of the spleen which may indicate a splenic sequestration crisis. Patients should also be assessed for signs of priapism as this is also an acute complication of SCD and is a medical emergency (Kalsi et el, 2002). Finally, dehydration may be present and the trigger for an acute episode, so assessing for signs of dehydration may prove useful in identifying the trigger. A full set of vital signs should also be documented, including a 12-lead electrocardiogram (ECG) to rule out any cardiac cause of the pain (JRCALC, 2009a).
It is known there are certain factors that can trigger an acute crisis so gaining a detailed history from the patients is important. Triggers can include:
Vaso-occlusive crisis
The most common problem for SCD sufferers is a vaso-occlusive crisis (VOC)—this is when the deoxygenated HbS polymerize, causing RBC sickling. Sickled RBC attach to vascular endothelium which can obstruct the lumen of a blood vessel. This in turn causes ischaemia and tissue infarction and can eventually result in organ failure (Figure 2). It is VOC which causes the classic presenting symptom of severe pain in SCD sufferers (White, 2005). This can occur suddenly and in almost any area of the body, and repeated episodes can cause irreversible damage to organs. Dactylitis occurs most commonly in children with VOC, this is when hands and feet become swollen and painful.
Acute chest syndrome
Acute chest syndrome is the second most common complication and causes up to 25% of deaths in patients with SCD (Platt et al, 1994). Acute chest syndrome occurs when vascular occlusion and inflammation affect areas of the bronchial tree. Acute chest syndrome should be suspected in any patient presenting with the clinical signs and symptoms of chest pain, cough, wheezing, tachypnoea and fever (Martin and Gunder, 2010). Still, very little is known regarding aetiology of acute chest syndrome (Bernard et al, 2007).
Early recognition of clinical signs and symptoms, early transport to the emergency department (ED) and a comprehensive handover will ensure treatment is initiated promptly on arrival at hospital.
Splenic sequestration crisis
Another potentially life-threatening consequence of SCD is a splenic sequestration crisis. It is mainly found in young children with SCA and occurs in approximately 10-30% of those with the disease (Harris, 2011). It is characterized by acute abdominal pain and enlargement of the spleen; caused by blood pooling within the organ, as the blood vessels leading out of the spleen have been blocked by sickling. If the enlargement is sudden, there can be a large decrease in the blood count and circulating blood volume, leading to potentially life-threatening complications (Table 2). Children with this condition will require urgent medical attention and possible blood transfusion.
| Cold/clammy |
| Left sided abdominal mass Pallor |
| Tachycardia |
| From: Crain and Gershel (2010) |
| Hypotension. |
Repeated splenic complications such as splenic infarctions can cause asplenia. This is significant because bacterial infection is the leading cause of death in children with SCD. During childhood, the spleen is often so badly damaged that it shrinks and no longer functions (autosplenectomy) (Mattei, 2011). For this reason, children under 5 will receive a prophylactic daily dose of penicillin (Cober and Stephanie, 2010).
Stroke
Stroke can be one of the most devastating and life altering complications of SCD, with an estimated 11% of patients with homozygous SCD (i.e, they have two copies of the SCT gene—HbSS), having a stroke by the age of 20 years (Ohene-Frempong et al, 1998). For this reason, it is important to observe closely for any neurological signs the patient may present with and also undertake a FAST (face; arm; speech; time) test. The stroke can be ischaemic or haemorrhagic in origin, although haemorrhagic strokes are most common in the 2029 age group (Sickle Cell Society, 2008). Children may also develop a silent cerebral infarct—this is when findings on an MRI suggest an infarction has taken place without the clinical neurological signs one would expect (Adekile et al, 2002).
Prehospital management
Guidance exists in several forms for those suffering from an acute sickle cell crisis. The Sickle Cell Society (2008) and JRCALC (2009a) both have specific guidance on the management of a sickle cell crisis which the prehospital practitioner can use. Management will be based primarily on the patient's presenting symptoms which will normally be severe pain—this accounts for 90% of hospital episodes. The patient may also have been given an individualized treatment plan outlining which type of SCD they suffer, along with guidance on treatment regimes that are most effective for them, based on previous exposure.
One problem highlighted in a report by Lucas et al (2008) into sickle cell care was the poor and often delayed clinical care for those patients presenting with acute complications of sickle cell disease. This emphasizes the importance of initiating treatment prior to ED arrival. JRCALC (2009a) recommend that the patient should be conveyed to their normal treatment centre unless any life-threatening conditions exist, in which case they should be taken to their nearest most appropriate receiving facility. JRCALC (2009a) also suggest that patients should not walk to the ambulance, as this may exacerbate the effects of hypoxia in the tissues. Many severe cases may also leave patients physically unable to walk.
Oxygen
Oxygen does not need to be routinely administered to adults unless the patient is presenting as hypoxaemic (O'Driscoll el al, 2008). JRCALC (2009b) recommends the administration of 2-6 litres per minute via nasal cannula, or 5-10 litres via a simple face mask, until a reliable Sp02 reading is obtained, then maintaining 94-98% or a normal range for the patient. Hypoxemia will aggravate RBC sickling, increasing symptom severity, so monitoring for any change in the patient's Sp02 and correcting hypoxemia early is important, particularly with acute chest syndrome as an increase in pain could have a detrimental effect on the patients breathing.
Fluid replacement
Dehydration may occur as the patient may have been unwell for some time before calling for assistance and it may also have triggered the crisis. Fluid replacement for dehydration is needed over hours rather than minutes and therefore is not going to be achieved in the average journey to hospital (Rees et al, 2003). Clinicians should aim to maintain radial pulse in line with JRCALC (2006) guidelines for fluid resuscitation in medical emergencies, unless there are other signs of poor central tissue perfusion. The Sickle Cell Society (2008) also recommends avoiding all unnecessary cannulation and fluid replacement as repeated venous access will damage vessels.
Pain management
There is no high level evidence on the prehospital management of acute pain with SCD. The management is based on expert opinion and what is currently known about hospital-based analgesia. Health professionals may have misguided perceptions that the patient is suffering from some form of addiction because certain pain behaviours are similar to symptoms of analgesic addiction. Based on this, they may also underestimate the level of pain that the patient is suffering (Elander et al, 2006).
Pain management is key in improving the patient's condition and relieving symptoms. Analgesia that has already been administered prior to arrival of the clinician should be taken into account when choosing the most appropriate pain management strategy.
‘Managing acute complications with sickle cell disease has to be a multi-disciplinary approach with prehospital clinicians playing a key role’
A stepwise approach to pain management should be used. For moderate pain, non-steroidal anti-inflammatory drugs (NSAIDs) such as ibuprofen and paracetamol may be effective; but more than likely, the patient will be presenting with severe pain, requiring a more potent analgesic. Pain needs to be relieved rapidly and the Sickle Cell Society (2008) state first dose of analgesia needs to be administered within 30 minutes and pain must be under control within 60 minutes. Guidance with specific or standardized drugs is not appropriate for the management of a crisis and the Sickle Cell Society (2008) also recommend that analgesic preference should be based on history of previous exposure, and also the patient's choice.
A prehospital clinician's choice of analgesics is somewhat limited in comparison with what ED has to offer and may not include analgesics which have previously been effective. However, this should not deter the practitioner from offering suitable analgesia prior to arrival at ED. Entonox can be administered if not contraindicated. Due to its rapid onset of action, it is ideally suited to relieving pain quickly, while other analgesia more suitable for prolonged use is sought. Entonox is not recommended for prolonged use in SCD due to the potential for anaemia and nerve damage to occur (Dart, 2004).
Morphine is a strong opioid analgesic, its onset time is slower in comparison to Entonox, taking 10-20 minutes for peak effect to be achieved but its duration of effect can last up to 4 hours. With severe pain, it is most commonly administered intravenously as this will provide quickest relief. However intravenous access may be difficult with SCD patients due to previous repeated venous access.
JRCALC (2009a) therefore recommends oral morphine administration or subcutaneous injection as these maybe more appropriate and easier to achieve, although reference should be sought from the patients personalized plan, if available, and analgesia delivered based on information given in the plan.
The Joint Formulary Committee (2011) and Rees et al (2003) also recommend the use of a NSAID at the same time as it may increase the effect of the morphine and allow lower doses to be used. This is supported by Jimenez-Andrade et al (2006) who found NSAIDs and opioids analgesics have a synergic effect on each other.
Conclusion
Managing acute complications with SCD has to be a multidisciplinary approach with prehospital clinicians playing a key role. Management will normally be based on presenting symptoms, with close attention being paid to potentially life-threatening complications.
It should be remembered that all patients with SCD will present differently and with varying symptoms, which is why a thorough assessment is vital. Patients may also have an individualized treatment plan which, where possible, should be followed.
A patient's perceived level of pain should not be underestimated and analgesia should be provided where clinically indicated to relieve the patient's pain. Where possible, the patient should be conveyed to their specialist centre where they are normally treated for further assessment.