
Despite advances in revascularisation strategies, dual antiplatelet therapy and intravenous anti-thrombotic therapies, cardiogenic shock complicating ST-segment elevation myocardial infarction (STEMI) remains an unresolved medical challenge (Werden et al, 2014)—and the leading cause of mortality in the STEMI population.
De Backer et al (2010) report that next to septic shock, cardiogenic shock is the most common reason for intensive care admissions. Hasdai et al (2000) comment that it is associated with more than 40% acute reduction in left ventricular function. Such a reduction in left ventricular function presents as haemodynamic instability, end-organ hypoperfusion and the development of multi-organ dysfunction syndrome, which may lead to death. Multi-organ dysfunction syndrome is characterised by more than one vital organ dysfunction and is associated with a systemic inflammation process (Parke et al, 2003).
Survival can be improved through the identification of high-risk patients and early recognition of clinical development with prompt implementation of preventative and treatment strategies. This article is part one of two and will focus on the defining characteristics of cardiogenic shock, its pathophysiology, clinical presentation and patient assessment.
Defining cardiogenic shock
Several definitions exist in clinical guidelines and randomised controlled trials. van Diepen et al (2017) describe cardiogenic shock as a primary cardiac disorder causing a reduction in cardiac output, resulting in clinical and biochemical signs of end-organ hypoperfusion, despite adequate circulating volume. Thiele et al (2015) outline a clinical spectrum that ranges from signs of mild hypoperfusion to profound shock with evidence of multi-organ dysfunction. In the most recent European Society of Cardiology (ESC) guidelines on the management of STEMI, Ibanez et al (2017) describe cardiogenic shock as persistent hypotension (systolic blood pressure (SBP) <90 mmHg) with evidence of end-organ hypoperfusion, which is commonly associated with extensive left ventricular dysfunction. Hollenberg et al (1999) comment that upon arrival in hospital, cardiogenic shock is confirmed once exclusion or correction of hypovolaemia, hypoxia and acidosis, has occurred and there is evidence of left ventricular dysfunction such as on the echocardiogram. The patient being transferred to a referral cardiac centre who is being maintained on inotropes and/or mechanical circulatory support (such as an intra-aortic balloon pump to maintain end-organ perfusion) and with a systolic blood pressure >90 mmHg, is also considered to be in cardiogenic shock (McMurray et al, 2012). Thiele et al (2015) outline the following diagnostic characteristics:
Menon et al (2000) demonstrated that cardiogenic shock can develop gradually, where the patient remains normotensive but has an increased heart rate and respiratory rate due to active compensatory mechanisms. This is known as a preshock state and aims to maintain end-organ perfusion. As compensatory mechanisms fail to maintain blood pressure and perfusion, the severity of shock progresses to a state where the patient requires inotropic and/or mechanical circulatory support in an attempt to preserve end-organ perfusion.
Causes
According to Reynolds and Hochman (2008), any cause of severe left ventricular or right ventricular dysfunction may lead to the development of cardiogenic shock. Acute myocardial infarction (AMI) causing left ventricular dysfunction is the most common cause of cardiogenic shock and accounts for 80% of cases (Hochman et al, 2000). Right ventricular involvement associated with an inferior STEMI causing cardiogenic shock, according to van Diepen et al (2017), accounts for 5.3% of myocardial infarction (MI)-induced cardiogenic shock. Mechanical complications associated with STEMI such as ventricular septal defect, cardiac wall rupture and acute mitral incompetence are less frequent causes. These complications are difficult to recognise in the prehospital setting and are diagnosed on echocardiography.
The reported incidence of mechanical causes are described by Hochman et al (1995) as acute mitral incompetence 8.3%, ventricular septal defect 4.6%, and cardiac wall rupture 1.7%. Non-MI causes include acute fulminant myocarditis, acute decompensated heart failure, arrhythmias, and myocardial contusion (Hollenberg et al, 1999).
Prevalence and prognosis
Despite advances in medical therapies and percutaneous coronary intervention (PCI), mortality rates according to Werden et al (2014) remain greater than 40%, particularly if multi-organ dysfunction syndrome is present. Goldberg et al (2009) report the incidence of cardiogenic shock complicating MI to be 6–10%, which Thiele et al (2010) translate into approximately 60 000–70 000 people in Europe per year. In describing the decade-long trends of incidence and hospital case mortality rates, Goldberg et al (2016) demonstrated that among the 5686 patients admitted with AMI, 3.7% developed cardiogenic shock after hospital admission with reported in-hospital mortality being 41.4%. Although a high in-hospital mortality rate is demonstrated, Goldberg et al (2016) found a decline in mortality rates from 47.1% in patients enrolled between 2001 and 2003, to 28.6% in those enrolled from 2009–2011. This decline in mortality can be attributed to an increase in the use of evidence-based cardiac medications such as dual antiplatelet therapy administered in the prehospital setting and direct transfer to primary PCI centres, as well as advances in PCI.
Integral to reducing mortality is the identification of those who are high risk of developing cardiogenic shock. Lindholm et al (2003) identified several patient characteristics that are deemed high risk for shock development. These include an anterior MI, a history of hypertension or diabetes mellitus, or patients with a history of left ventricular dysfunction. Other characteristics include the presence of left bundle branch block on the electrocardiogram (ECG), a prior acute coronary syndrome event or the presence of multi-vessel coronary artery disease. Goldberg et al (2016) identified that those who developed cardiogenic shock as inpatients were significantly older, were more likely to have a do-not-resuscitate order in their hospital record or were transported to a referral hospital by ambulance. Clinical characteristics include a higher heart rate, lower blood pressure and a raised serum glucose on admission. Factors associated with an increased mortality rate include older age, anoxic brain damage, lower left ventricular ejection fraction, lower systolic blood pressure, administration of vasopressor therapy, and worse renal function (Thiele et al, 2015). The identification of high-risk patients is achieved by undertaking a targeted history and a thorough patient assessment.
Short-term patient outcomes are related to the severity of haemodynamic instability and end-organ involvement. For those who survive, Shah et al (2016) demonstrated that 1-year all-cause and heart failure rehospitalisation rates were 59% and 33%, respectively. In relation to quality of life and functional capacity at 1 year, quality of life was quantified using the New York Heart Association (NYHA) classification, where 43% of patients were classified from class II–IV, with 20–30% experiencing challenges in physical or psychological self-care ability (Sleeper et al, 2005; Thiele et al, 2013).
Pathophysiology
The pathophysiology underpinning cardiogenic shock is complex and multifactorial. For those experiencing a STEMI, shock develops when more than 40% of the left ventricular muscle is compromised, which decreases the strength of contractility. Alonso et al (1973) illustrated that in 22 patients who died from cardiogenic shock complicating MI, the infarction zone affected approximately 51% of left ventricular mass compared with 23% ventricular mass in 10 patients who died from myocardial infarction without shock.
The decrease in left ventricular function causes stroke volume and cardiac output to decline, which decreases blood pressure and limits adequate end-organ perfusion. As the cardiac output decreases, Thiele et al (2015) describe a fall in coronary perfusion pressure exacerbating myocardial injury and ischaemia, where myocardial cells switch from aerobic to anaerobic metabolism. This results in a vicious spiral causing further reduction in contractility, stroke volume and cardiac output.
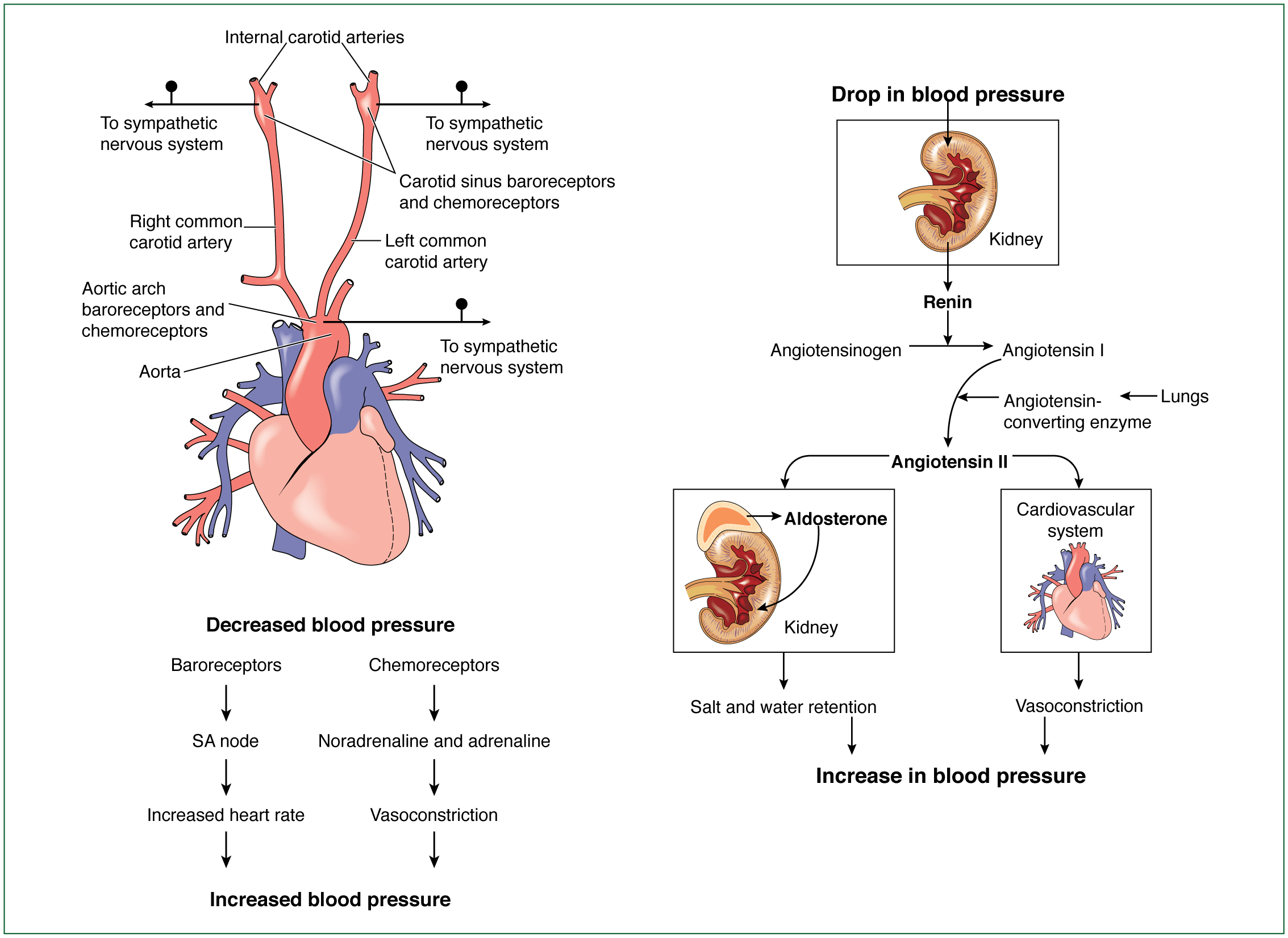
In response to a suboptimal cardiac output, compensatory mechanisms—namely the sympathetic and renin–angiotensin–aldosterone system (RAAS)— are activated as a means of protecting end-organ perfusion by increasing contractility and peripheral blood flow (Reynolds and Hochman, 2008). The sympathetic nervous system promotes the release of catecholamines, adrenaline and noradrenaline. These catecholamines are beta-receptor stimulants, promoting an increase in heart rate, contractility and selective vasoconstriction as a means of optimising cardiac output. The RAAS stimulates the production of angiotensin II and aldosterone, which increase afterload by vasoconstriction and increase preload by conserving sodium and water respectively.
Although the objective is to maintain end-organ perfusion, Aymong et al (2007) comment that these compensatory mechanisms increase myocardial oxygen demand in the presence of limited coronary oxygen supply, exacerbating myocardial injury and ischaemia. In addition, an enhanced preload and afterload increase the failing left ventricular workload.
In response to acute myocardial injury, a systemic inflammatory response is triggered. This causes a release of excess nitric oxide from endothelial cells within smooth muscle, resulting in microcirculatory vasodilation and interference in catecholamine function.
Hochman et al (2003) outline the negative properties of nitric oxide in reducing contractility and its cardiotoxicity. Reynolds and Hochman (2008) describe the impact of this vasodilatory response as further inhibiting end-organ perfusion, in particular, to the intestinal tract. The vasodilation in the intestinal tract enables transmigration of bacteria, potentially increasing the risk of sepsis. In a review of the ‖SHould we emergently revascularise Occluded Coronaries for cardiogenic shocK (SHOCK)‗ data, Kohsaka et al (2005) illustrated that sepsis was suspected in 18% of the study population, of whom 74% developed positive bacterial cultures. With a systemic inflammatory response, there is release of cytokines, tumour necrosis factor–α and interleukins–6, which further depress myocardial perfusion and contractility.
Thiele et al (2015) describe the net effects of the compensatory mechanisms and systemic inflammatory response as microcirculatory dysfunction, further myocardial ischaemia and injury, and increased afterload and preload, which further burden the failing left ventricle. The initial myocardial injury, stimulation of the compensatory mechanisms and the systemic inflammatory response create a self-perpetuating circle that results in the onset of haemodynamic instability and end-organ hypoperfusion.
Clinical presentation
The signs and symptoms associated with cardiogenic shock result from left ventricular dysfunction and activation of compensatory mechanisms. According to Vincent and De Backer (2013), clinical presentation is characterised by persistent hypotension where the systolic blood pressure is <90 mmHg or the mean arterial pressure is <70 mmHg with associated tachycardia that is unresponsive to fluid replacement. The associated tachycardia is a result of the compensatory sympathetic drive. Compensatory mechanisms lead to increased respiratory rate and workload, cool peripheries and clammy skin with delayed capillary refill.
Aymong et al (2007) comment that the organs most prone to the effects of decreased perfusion are the kidney and the brain. This is evidenced by a reduced urine output of <0.5 mls/kg/hour. From a neurological perspective, altered mental status occurs which progresses to obtundation and coma. As a consequence of decreased cardiac output and increased ventricular wall stress, pulmonary congestion may occur. On examination, this is confirmed by tachypnoea, respiratory distress and a decrease in oxygen saturations to <90%.
Diagnosis
In the prehospital setting, cardiogenic shock is suspected based on the presenting history, clinical presentation, physical examination findings and a 12-lead ECG confirming the presence of a STEMI or a left bundle branch block associated with ischaemic chest pain. Upon arrival to hospital, echocardiography and serum laboratory tests play an essential role in confirming the diagnosis along with prehospital findings. Hollenberg et al (1999) comment that integral to diagnosis is the exclusion of other causes of hypotension such as hypovolaemia, sepsis, pulmonary embolism, cardiac tamponade and arrhythmias.
Upon arrival at the scene, initial primary and secondary survey is needed, especially if the patient is in decompensated shock. A full cardiovascular assessment in conjunction with a targeted history is then essential for identifying a provisional diagnosis of cardiogenic shock. A targeted history involves using SAMPLER:
This provides clues regarding the cause of the clinical presentation, as well as insight into patients who are at high risk of developing this form of shock. According to Vincent and De Backer (2013), a full cardiovascular examination includes assessment of skin colour and temperature, jugular venous pressure and peripheral oedema, as well as auscultation of the heart and lungs. Findings from the assessment may include cool clammy skin, weak or thready pulses on palpation, mottled extremities and altered orientation. The jugular vein may be distended and, on auscultation, a 3rd and/or 4th heart sound may be present. From a respiratory perspective, the patient may demonstrate increased respiratory rate and work of breathing, decreased oxygen saturation and pulmonary rales on auscultation.
A 12-lead ECG would demonstrate evidence of STEMI. Ibanez et al (2017) outline the ECG criteria for STEMI as being 1 mm ST segment elevation in all leads except V2–V3 where the following applies:
Hochman et al (2000) demonstrated that in those experiencing cardiogenic shock, 55% experienced an anterior STEMI, 46% an inferior STEMI, 19% posterior, 32% a lateral MI and 11% an apical MI.
Upon arrival in hospital, echocardiography has a pivotal role in the diagnosis of cardiogenic shock. Ibanez et al (2017) recommend a bedside study in assessing ventricular and valve function. In addition, assessment can confirm or rule out mechanical complications associated with STEMI such as ventricular septal defect, acute mitral incompetence or pericardial effusion. The echocardiogram may be repeated throughout the treatment period as a means of guiding and evaluating therapeutic strategies. Repeat studies may demonstrate improvement or deterioration in ventricular function, presence of pericardial effusions, determination of valvular function and wall motion abnormalities. The frequency of echocardiography studies is guided by patients’ haemodynamic and clinical status.
As part of the patient assessment, routine bloods and a chest x-ray are obtained. Routine bloods such as full blood count, renal and liver function, cardiac markers, arterial blood gases and lactate are reserved. In the setting of a STEMI, cardiac troponin will be increased, indicating myocardial cell necrosis. Arterial blood gases measure arterial oxygenation and ventilation, as well as provide information on the metabolic and respiratory acid base status. With end-organ hypoperfusion, acute kidney and liver injury are evidenced by a rise in the serum creatinine and liver enzymes such as serum aspartate aminotransferase, alanine aminotransferase, bilirubin and lactate dehydrogenase levels. A renal function test will provide information on electrolyte balance. A chest x-ray provides information on the size of the heart and the degree of pulmonary congestion. For those patients who are ventilated, it will confirm position of the endotracheal tube and the position of supportive devices such as temporary pacing wires and mechanical circulatory support such as intra-aortic balloon catheter. In relation to invasive haemodynamic monitoring, Ibanez et al (2017) state that insertion of a pulmonary floatation catheter to measure intra-cardiac pressures, cardiac output and cardiac index is not routinely mandated but may be indicated in cases of shock associated with unknown cause or in patients who are unresponsive to therapeutic modalities as a diagnostic tool.
Once the diagnosis of shock is established, therapeutic strategies are aimed at identifying and correcting the underlying cause with the overall aim of preserving myocardial and end-organ function.
Conclusion
Cardiogenic shock complicating STEMI is a potentially life-limiting condition with mortality rates exceeding 40%. Clinical presentation comprises persistent hypotension despite adequate circulating volume and evidence of end-organ hypoperfusion. Signs of end-organ hypoperfusion include cold clammy skin, delayed capillary refill, evidence of pulmonary congestion, decreased urinary output and, for some, altered mentation. The potential for death can be reduced with identification of those who are at high risk and early recognition of its development. The pathophysiology is complex and multifactorial with signs and symptoms attributed to a decrease in ventricular function and activation of compensatory mechanisms. Patient assessment focuses on the extent of haemodynamic compromise and end-organ hypoperfusion. Integral to assessment is the identification of a potential reversible cause. Part 2 of this series will discuss the role of evidence-based treatments in this patient population. JPP
A version of this article was first published in the January 2019 issue of the British Journal of Cardiac Nursing.