
Globally, major trauma is the leading cause of death among children and young adults under the age of 40 (Krug et al, 2000). While there are no current complete data on the incidence of major trauma and mortality rates in England, the most recent available data collected by the Trauma Audit and Research Network (TARN) estimate that there were 20 000 incidents of major trauma in 2007, with at least 5400 of these leading to death (National Audit Office (NAO), 2010). However, these data are over a decade old and may not be representative of current incidence. Advances in pre-hospital care, such as the introduction of tranexamic acid (TXA), and implementations of systematic trauma quality improvement systems over the past decade have been associated with reduced mortality rates (O'Reilly et al, 2013).
Within the UK, 12 ambulance service trusts respond to 999 and 111 calls, collectively receiving 6.15 million cases in 2008-09, which resulted in an emergency response (NAO, 2010). It is estimated that 20 000 of these calls are regarding major trauma, suggesting that major trauma is a relatively small proportion of a paramedic's workload (NAO, 2010). However, owing to the nature and severity of injuries, pre-hospital interventions to rectify life-threatening problems are essential to improve patient outcomes.
One complication of major trauma is major haemorrhage (O'Reilly et al, 2013). Death by haemorrhage following major trauma is consistently the second leading cause of death following severe central nervous system (CNS) injury. Haemorrhage accounts for 30–40% of trauma mortality, with 33–56% of these deaths occurring in the pre-hospital setting (Kauvar et al, 2006). However, major haemorrhage also remains the highest preventable cause of death in trauma (Bouglé et al, 2013); therefore, bleeding patients are a clinically significant subgroup of the trauma population. The immediate priorities for clinicians attending trauma patients were revised in 2006, identifying that correcting major haemorrhage should come before airway management, cervical spine stabilisation, breathing, and circulation (Hodgetts et al, 2006).
It has been recognised that a proportion of patients with trauma arrive in emergency departments with an established clotting disorder known as acute traumatic coagulopathy (ATC) or trauma-induced coagulopathy (TIC). This pathology is associated with an accelerated rate of bleeding, and a four-fold increase in mortality rate (Brohi, 2003). As advances in pre-hospital medicine are introduced to treat this pathology, such as TXA, it is important that paramedics understand how current and new treatments interact with the haemostatic response and benefit the patient.
This is the first of two articles discussing pre-hospital major haemorrhage. The current article explores the physiological systems deranged by haemorrhage and set out the pathophysiology of these systems. It then discusses the research covering advances in the understanding of how deranged coagulation occurs.
Physiological pathways affected by major haemorrhage
Bleeding within the body affects two principal physiological systems:
Haemostasis
Any vascular damage triggers the body's normal haemostatic response; an interaction between blood coagulation factors, platelets, and the vessel wall. To ensure minimal blood loss, a rapid and efficient mechanism to generate a thrombus is essential.
Nonetheless, the over-formation of thrombus can be pathogenic and this is seen with the morbidity of both venous thromboembolism, causing pulmonary emboli, as well as stroke and limb ischaemia secondary to arterial thrombus formation. Therefore, there is a delicate balance within the haemostatic system between procoagulant and anticoagulant mechanisms and the breakdown of clots, i.e. fibrinolysis (Hoffbrand and Moss, 2016).
The normal haemostatic response consists of five major components:
The operation of these components is illustrated in Figure 1. In summary, vessel injury causes activation of the coagulation cascade illustrated in Figure 2, by two routes known as the intrinsic and extrinsic pathways. These pathways follow the initial formation of a platelet plug. The intrinsic pathway, or contact activation pathway, is activated by platelets in traumatic vessel injury. Research has suggested that this pathway is less significant in the overall generation of fibrin via thrombin, as deficiencies of clotting factors do not lead to major bleeding disorders (Palta et al, 2014).
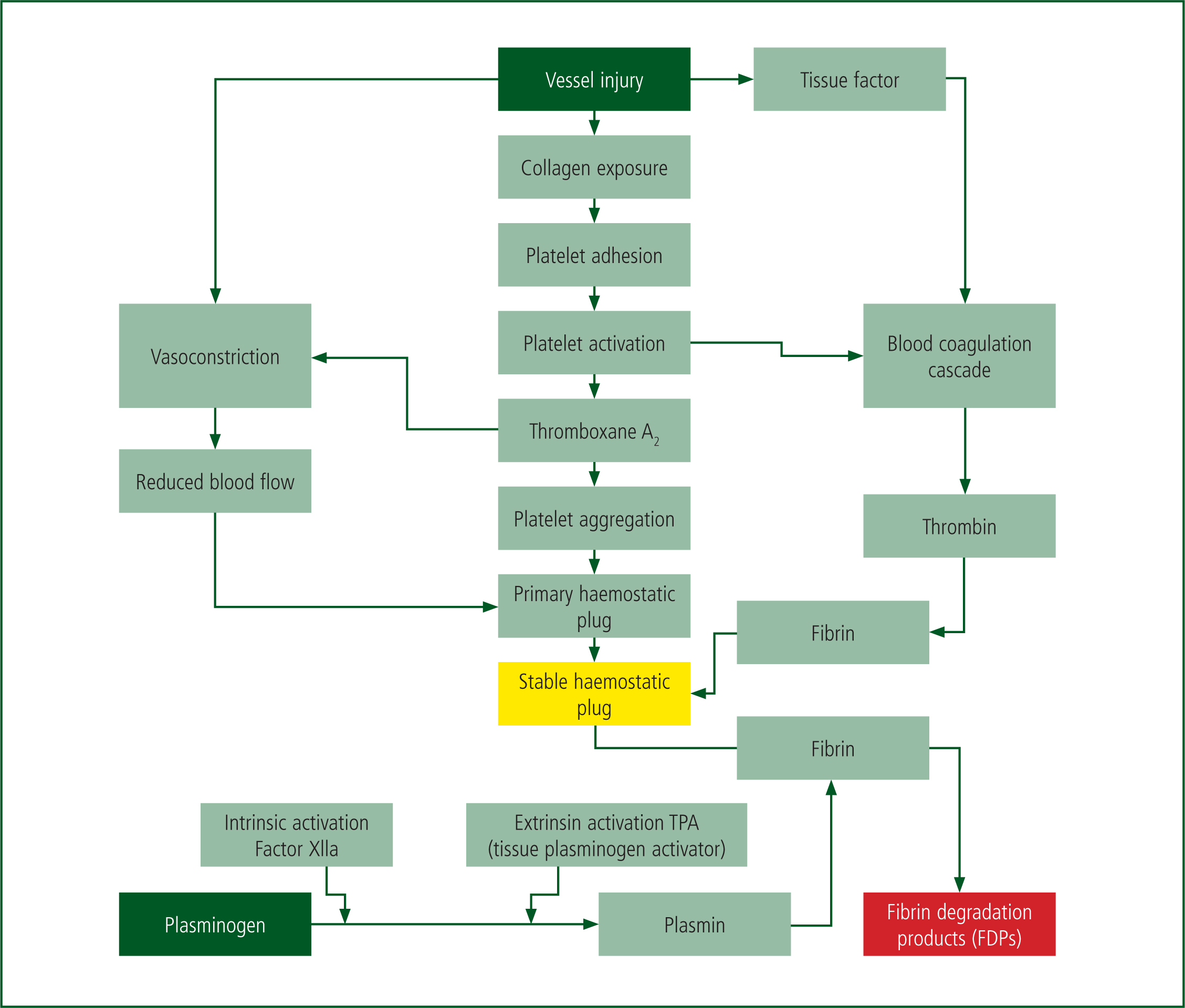
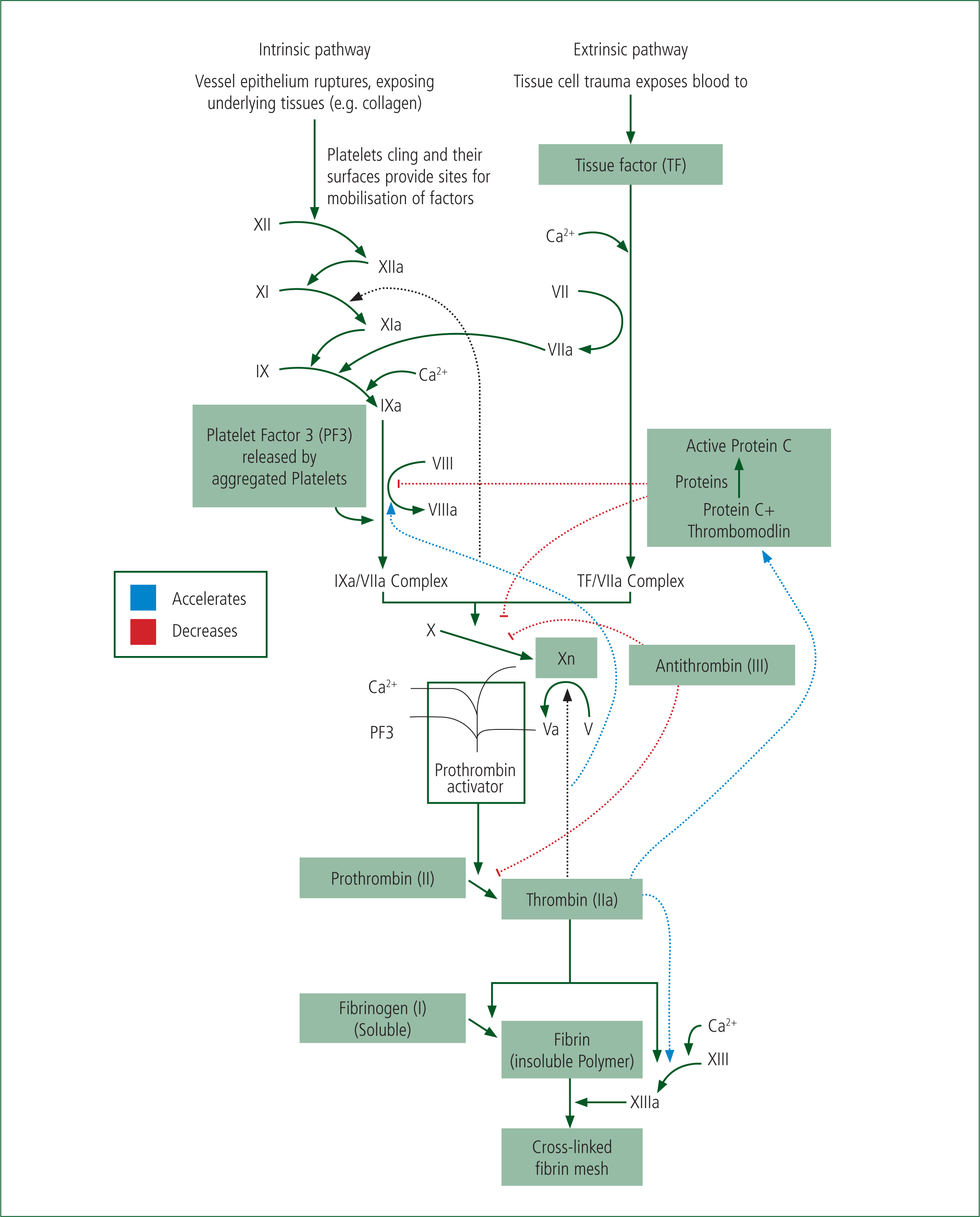
The extrinsic pathway, activated by tissue factor, which becomes exposed on stromal cells during trauma leads to the rapid activation of clotting factor X to Xa, and the subsequent production of thrombin from prothrombin. The production of thrombin is key as it catalyses the production of fibrin from fibrinogen, and thereby the stabilisation and formation of a clot—a thrombus (Hoffbrand and Moss, 2016).
Knowledge of this pathway allows for paramedics to understand how anticoagulant drugs, ‘blood thinners’, exert their effects. For example, Warfarin, a vitamin-K antagonist, acts by reducing the production of clotting factors which rely on vitamin K for their production (II, VII, IX, X). Aspirin and clopidogrel act against cell surface receptors which are located in platelets. Newer anti-coagulants such as rivaroxiban and apixaban act by directly inhibiting Factor X and its activation. Heparins, including the low-molecular weight versions, such as dalteparin, act by activating one of the physiological anticoagulants, Factor III—antithrombin (Hoffbrand and Moss, 2016).
As a clot is formed, fibrinolysis/clot break-down is also occurring to ensure the clot does not become pathological. During fibrinolysis, plasminogen activators (t-PA) from injured blood vessels convert plasminogen to plasmin. Plasmin then binds to the fibrin clot via a lysine binding site, and this breaks down the fibrin clot into its degradation products (Figure 3).
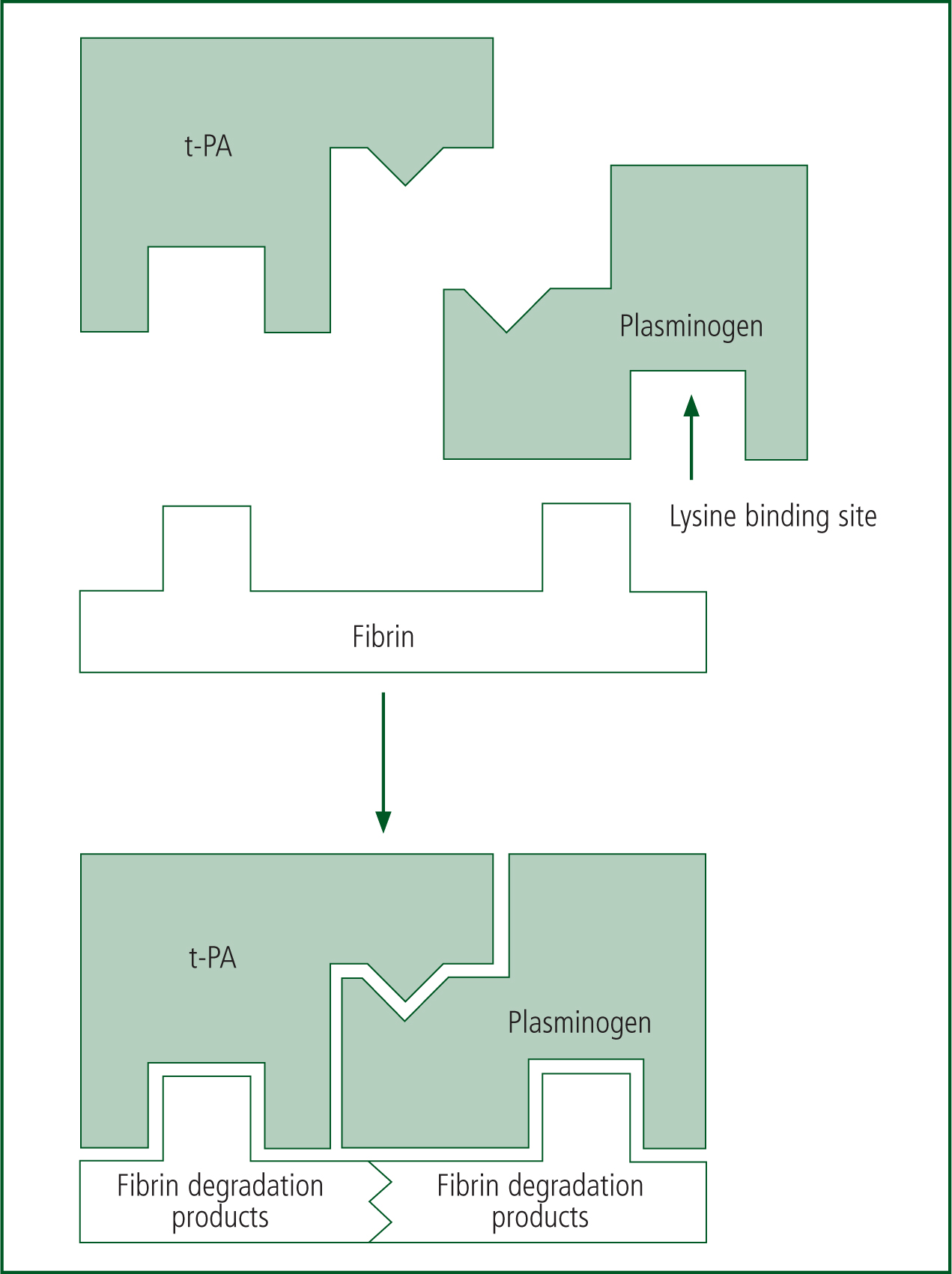
In summary, during a major haemorrhage, the damage caused to vessels activates a complex physiological pathway. In the healthy patient, this normally generates a fast response (local vasoconstriction and platelet plug formation), which is usually followed by a series of molecular changes to stabilise the platelet plug and form a thrombus—this can effectively seal off the bleeding point and act to prevent blood loss.
Blood pressure
The second pathway which is affected by major haemorrhage is the maintenance of tissue perfusion via a systolic blood pressure. The maintenance of tissue perfusion is the fundamental function of the heart. Tissue perfusion allows the delivery of oxygen and metabolic nutrients to cells and waste product removal—this allows for the maintenance of the cellular processes which underpin life.
The physiology supporting this basic function of the heart has been extensively characterised, at varying levels. At the ‘beat-to-beat’ level, the cardiac output (CO) is simply a function of heart rate (HR) and the volume of blood pumped, stroke volume (SV); this can be summarised in the equation CO=HRxSV. This equation illustrates two key variables in supporting blood pressure: HR and the volume of circulating blood returned to be pumped, SV.
However, given Laplace's law from physics (Hopkins, 1966), it is known that in any cylindrical system, resistance of the cylinder (i.e. blood vessels) is a key determinant in the pressure within the system. The equation which gives blood pressure (BP) is BP=COxPR (peripheral resistance) (Boron and Boulpaep, 2016).
As CO is influenced by SV, the above equations are only accurate given SV remains constant. The three factors affecting SV include:
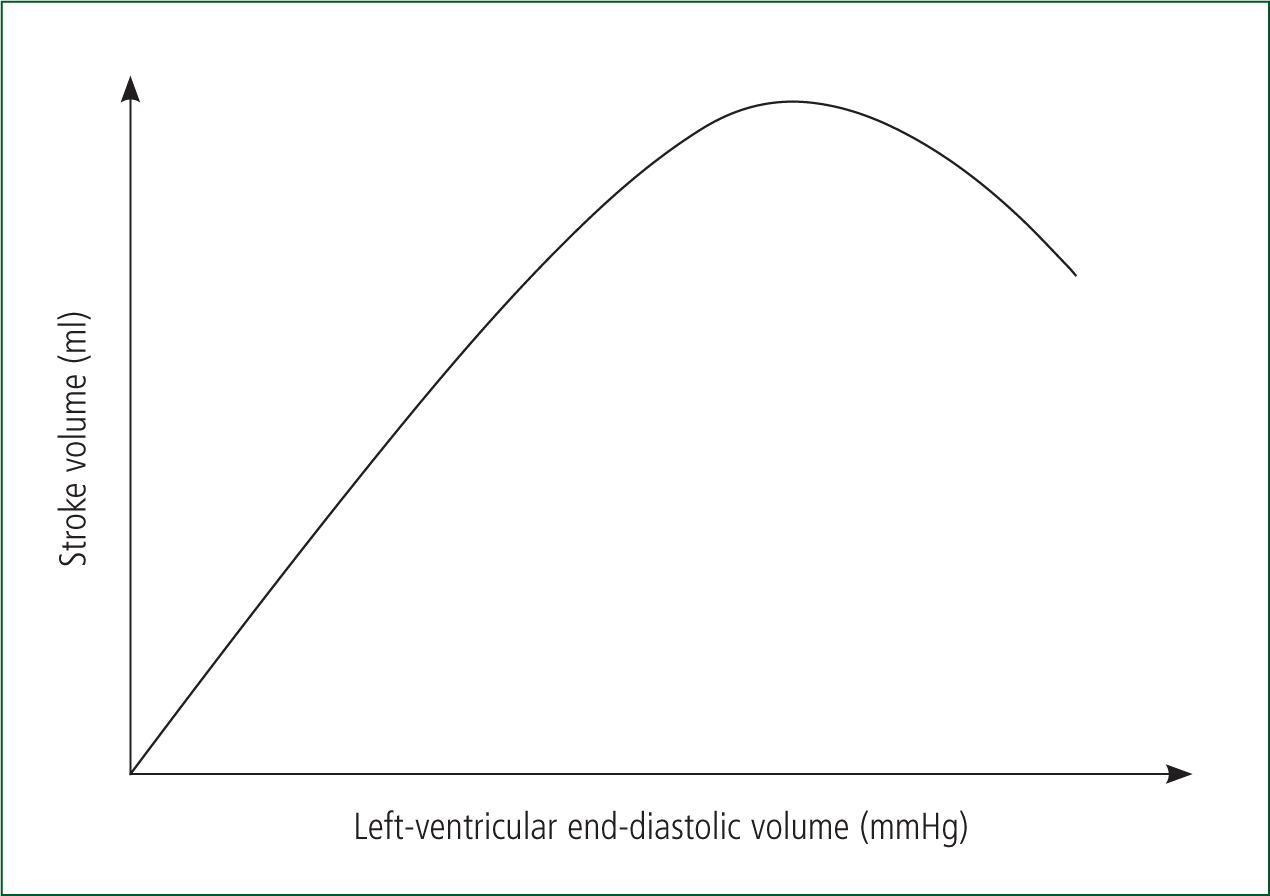
The relationship between preload and contractility has been demonstrated experimentally by Starling and Frank (Sequeira and van der Velden, 2015). It has been shown that an increased preload causes an increase in ventricular fibre length, resulting in an increased ‘tension’ within the myocardium. This then increases cardiac output, see the Frank-Starling curve in Figure 4. As venous return is the most important determinant of preload, SV and consequently CO are directly related to venous return (Gutierrez et al, 2013).
Regulation of blood pressure is exerted by systems which act to modulate CO or PR, these are principally under neuronal and hormonal control. Neuronal control acts via sensors, baroreceptors, located in the walls of major vessels (e.g. carotid sinus) which signal to both the brainstem (medulla) and spinal cord via the Vagus nerve. The vasomotor centre within the cord coordinates responses which acts via modulation of peripheral resistance, by vasoconstriction or dilation via the sympathetic and parasympathetic neuronal systems.
Vasoconstriction causes increased PR and raises blood pressure rapidly, while vasodilation decreases PR and lowers blood pressure. These immediate effects are allied with a consequent increase/decrease in the blood returned to the heart (preload) and also affect SV. It is also important to note that at the level of the brainstem, hypoxia is sensed via changes in the acid-base status of the blood. This in turn can cause increased sympathetic outflow centrally and raise blood pressure via increased HR and vasoconstriction to maintain SV (Boron and Boulpaep, 2016).
The second regulatory system is hormonal (endocrine) control. This is a complex system which mainly acts via altering blood volume and subsequently SV. The renin–angiotensin system acts via sensing renal perfusion. The kidneys release renin when renal perfusion is low and this then catalyses the conversion of angiotensinogen to angiotensin I (Ang I). Ang I is converted to angiotensin II (Ang II) by angiotensin-converting enzyme (ACE) in the lungs. Ang II causes arterial vasoconstriction, raising PR and increasing blood pressure. This ‘fast’ response is accompanied by a medium-term action to increase resorption of sodium at the kidneys, via aldosterone, which, as water follows sodium, increases blood volume (Boron and Boulpaep, 2016).
Pathophysiology of major haemorrhage
Having understood the systems mediating the normal physiology of haemostasis and blood pressure control, we can see how these are deranged when subjected to a major haemorrhage.
Significant blood loss can cause hypovolaemia, and if associated with a drop in blood pressure, can escalate to hypovolaemic shock—a syndrome causing inadequate perfusion of tissues. Paramedics should have high suspicion for hypovolaemia in bleeding patients, as experimental evidence has shown that the body can withstand up to 30% loss of circulating volume before the mean arterial BP is affected (Nolan and Pullinger, 2014).
Hypovolaemia
In hypovolaemia, the pathophysiology can be explained as a decrease in venous return (preload) causing a decreased SV and consequently decreased CO. At losses of more than 30% circulating volume, many of the physiological compensatory mechanisms can be seen. These can simply be understood in the framework of Le Chatelier's principle (Dainith, 2008) which applies to much of the physiology—the altered system will attempt to correct itself to the normal resting state.
As such, the compensatory responses follow the physiology outlined via altering CO or PR. The ‘fight-or-flight’ response comes first, acting over seconds to minutes. The decrease in blood pressure is detected by baroreceptors, in turn causing an increased sympathetic outflow and decreased parasympathetic outflow. This increases HR and PR, and therefore CO and BP. The increase in PR by arteriolar vasoconstriction occurs mainly in the periphery (via alpha adrenergic receptors), while the brain and coronary circulations are relatively protected (Hall and Guyton, 2015).
Compensatory responses acting over minutes to an hour are mediated by hormonal responses: the production of Ang II, adrenaline in the circulation and the production of aldosterone as discussed earlier. In addition, a process of fluid shifts across cellular components causes ‘internal transfusion’. As blood pressure falls, the hydrostatic forces acting between the intravascular and interstitial spaces are altered. Hydrostatic forces within the vasculature drop while the colloid osmotic pressure remains broadly the same. This causes a fluid shift from the interstitial fluid to the intravascular space.
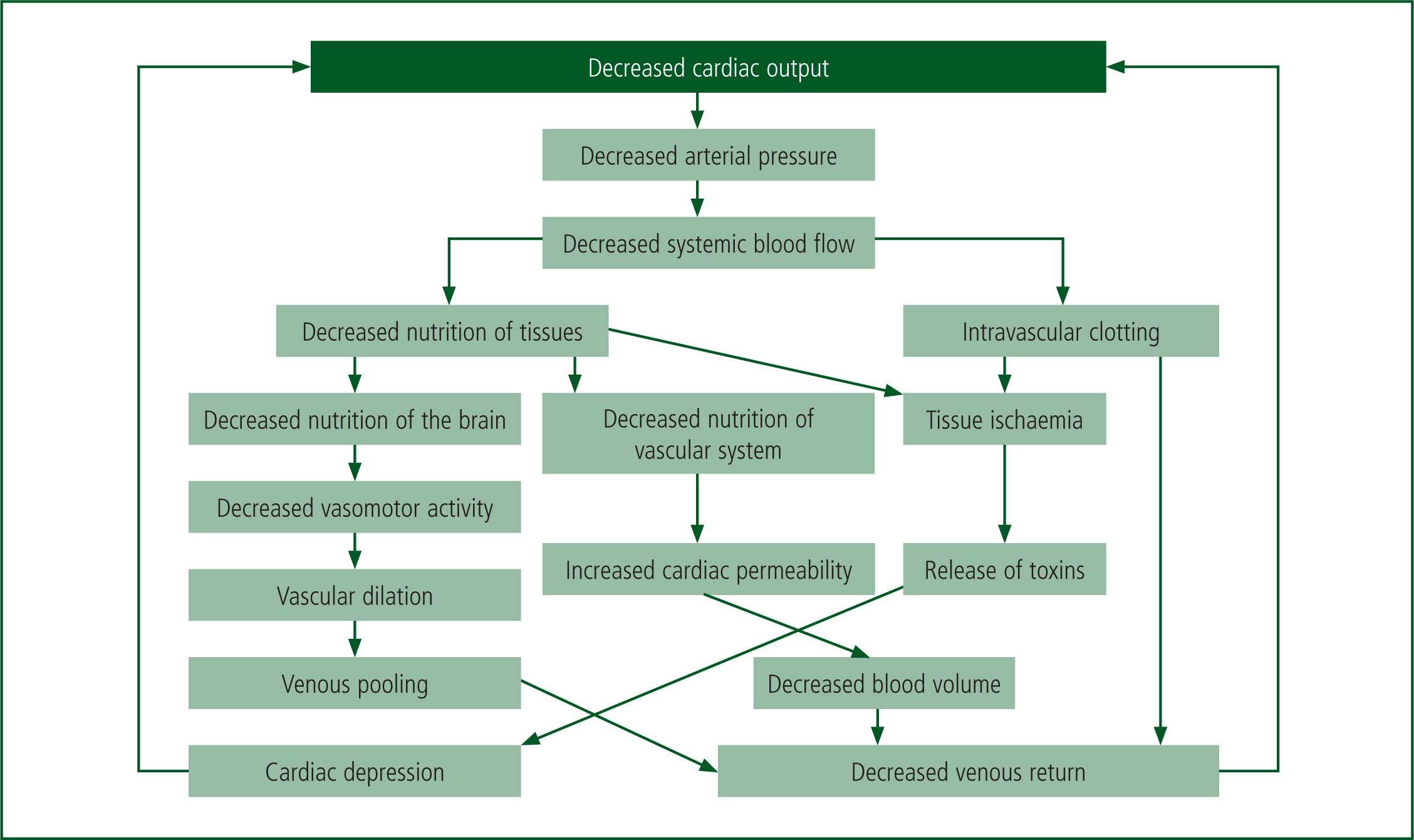
Hypovolaemic shock can be sub-classed into compensated or decompensated shock. Compensated shock occurs when the blood pressure is decreasing while the heart rate remains stable, decompensated shock is occurring where HR decreases with blood pressure. At this stage, sympathetic stimulation is lost and HR decreases, as does PR with vasodilation (Bonanno, 2011). This forms a pathological progressive positive feedback loop, which in turn leads to tissue ischaemia and ultimately death, as shown in Figure 5 (Hall and Guyton, 2015).
Coagulation
As noted in Figure 5, a factor in the pathophysiology of major haemorrhage is a derangement in coagulation. However, the pathophysiology here has only recently been understood. Brohi et al (2003) studied measures of clotting function (prothrombin time (PT), activated partial thromboplastin (aPTT), thrombin time (TT)) in 1088 trauma patients with hypovolaemic shock. These patients had only been subjected to minimal pre-hospital fluid therapy, to exclude dilution as a confounding variable, and the type of fluid therapy did not correlate with clotting function.
However, 25% of all of the patients had an established clotting disorder on arrival to the emergency departments (ED). Brohi et al (2003) concluded that a specific haemorrhage-related pathophysiology must be occurring, and named this acute traumatic coagulopathy (ATC). The consequences of a failure to clot are clear: increased blood loss and continuing hypovolaemia, which have been shown to affect mortality. In a study of over 5000 trauma patients, the clotting status was shown to be predictive of mortality (Frith et al, 2010).
Further research has begun to unravel the pathophysiological mechanisms at play in ATC, though undoubtedly the full picture is yet to be elucidated. Two factors have been identified as key:
Experimental rodent models of ATC suggested that during ATC, an excessive production of pC was key (Chesebro et al, 2009). The proposed mechanism is that thrombin switches its role from pro-coagulant, and cleaving fibrinogen, to anti-coagulant via activating thrombomodulin which in turn activates pC, which itself inhibits clotting factors V and VIII (Figure 2).
The reason for this switch is postulated that ischaemic tissue acts in an anti-coagulant manner to allow blood flow to relieve the ischaemia. Some evidence for this mechanism has been found. In a cohort of 300 trauma patients, increased levels of activated pC were found and these were correlated to transfusion requirements (Davenport et al, 2011).
The second factor that is seen in ATC is hyperfibrinolysis via the loss of fibrinogen (Rourke et al, 2012), and while this finding is empirically supported by cohort studies (Cotton et al, 2012), a precise mechanism has yet to be determined. Fibrinolysis can be analysed by thromboelastography (TEG)—a viscoelastic haemostatic assay that measures all phases of haemostasis in whole blood. The standard measurement for fibrinolysis is the clot lysis at 30 minutes after maximum clot strength (LY30). An LY30 measurement of 3% or greater defines clinically relevant hyperfibrinolysis. This is strongly associated with the requirement for massive transfusion and increased mortality in haemorrhaging trauma patients (Chapman, 2013). Hyperfibrinolysis (i.e. those with LY30 >3%) has been predicted to occur in 2–34% of trauma patients (Cotton et al, 2012), representing a critical indication for the treatment of fibrinolysis to reduce the risk of mortality and massive transfusions.
In summary, research continues to illustrate the complex molecular pathophysiology driven by the gross physiological insult caused by blood loss. Further understanding of these pathways may generate targets and strategies which could be applied to patient care.
Conclusion
The current article has set out the physiological systems of blood pressure control and haemostasis; explained their derangement in major haemorrhage; and discussed research covering advances in the understanding of trauma-induced coagulopathy. Paramedics may now apply this knowledge to understand the physiological processes when treating haemorrhagic patients.
A second article will discuss the assessment and management of major haemorrhage from an evidence-base point of view, linking this back to the pathophysiology set out in this article.
Avenues for further research include more preclinical research into the molecular pathophysiology of acute traumatic coagulopathy. These may yield new therapies which could be applied in the pre-hospital setting.