
Understanding the nature of drug actions that can occur in the body allows the paramedic to make informed decisions regarding medicines that may be offered, the likely outcomes and any possible risks. Despite often being considered a complex subject, this article presents a set of principles that are applicable to almost all drugs along with relevant case studies to support theory and provide context where appropriate.
Drug action
Many drugs exert their effect on the body via an action on cells. The net effect of this can be to:
Drugs can do this in a number of ways, but the most common methods are to chemically bind to the surface of a cell or, in the case of steroids, simply pass through the cell wall. Before discussing these methods in detail, we must acknowledge that these processes occur naturally in the body; our pharmacological interventions simply mimic the endogenous neurotransmitter or hormone. The importance of these endogenous chemicals cannot be understated. Indeed, before acetylcholine was the first neurotransmitter to be identified (Dale, 1914), synapses were widely considered to occur via an electrical process. The matter was largely put to rest after Loewi (1921) carried out his well known frog experiment which initially came to him in a dream. He stimulated the heart of a frog with electrical impulses and had it pump a small amount of nutrient solution. When the fluid was transferred to another heart, it operated similarly, proving that chemical substances convey nerve signals to organs.
G-protein coupling
Widely regarded as the largest family of drug targets, G-protein coupled receptors (GPCRs) frequently reveal the way in which drugs interact (Zhao et al, 2016).
When a drug binds to a receptor site, the physiochemical properties of that cell change; this is known as a conformational change. Next, the alpha sub unit of the G-protein dissociates and activates an enzyme which catalyses a further chemical reaction. The subsequent product is a second messenger molecule able to send signals to the body from within the cell (Figure 1). Taking adrenaline as an example, the second messenger signals could increase blood flow to skeletal muscle, increase glucose production, cause peripheral vasoconstriction or even bronchodilation. The effect would be largely dependent on dose and route. In this example, the second messenger molecule would be cyclic adenosine monophosphate (cAMP). It is important to note that cAMP is an intracellular signal transducer and does not itself represent a body that exits the cell.
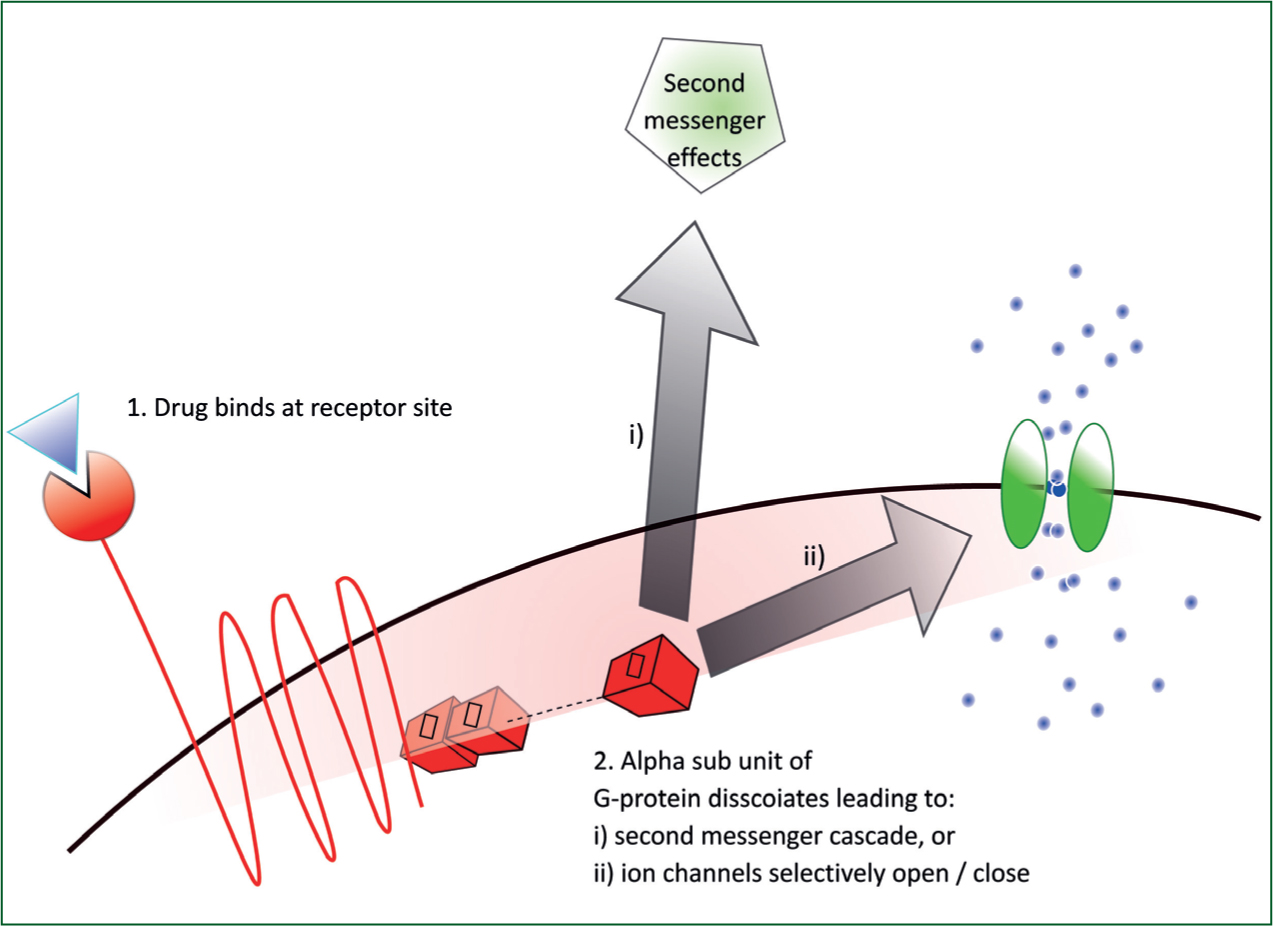
Alternatively, once dissociated, the alpha sub unit could block or open specific ion channels (Figure 1). The result here would be to prevent depolarisation, prolong repolarisation, raise or lower threshold potentials. The affects would depend on the location of these particular cells and the method of administration. A good example here would be lidocaine: local administration would block sodium channels and prevent (slow) depolarisation of nerve cells and hence provide local anaesthesia. However, when administered intravenously (IV), lidocaine blocks myocardial sodium channels and depresses myocardial contractility in ventricular tachycardia (VT) (Mccann, 2000).
Before moving on, we should consider the strength of the interaction between drug and receptor site and the concentration (amount of drug available). For a therapeutic result, the dose should be sufficient that a significant number of receptor sites are exposed to the drug; too little and there will be little or no physiological action, too much and we risk toxicity or overdose.
When a number of drugs, hormones or neurotransmitters are available to a receptor site, it is not simply a case of ‘first come, first served’; the receptor site will selectively bind to the molecule with the most preferable chemical properties. These will be a specific combination of chemical properties including structure, polarity (as well as how the charge may be distributed across the molecule), pH, solubility, etc. These properties refer to the affinity between drug and receptor, allowing for a more favourable drug to present itself as well as for displacement due to a greater affinity. A good example of this is Naloxone which is known as a competitive antagonist; since it has a greater affinity to opiate receptor sites, it will displace drugs (e.g. heroin, morphine) which are already bound to receptor sites. The same chemical properties mentioned are also responsible for the selectivity of a drug: that is whether a drug will prefer a particular receptor site or will happily bind to a number of different subtypes. Propranolol, a non-selective beta blocker which may be prescribed for arrhythmias, should be introduced cautiously in patients with a history of obstructive airways disease. Such patients are likely to be on beta-agonist inhalers (salbutamol) and in the presence of propranolol, the beta 2 receptor sites may well be blocked when the inhaler is required.
Steroid hormone receptors
Where G-protein receptor coupling occurs on the surface of a cell, steroids possess a unique ability to bind within cells. Steroids are able to diffuse across the plasma membrane of a cell due to their lipophilic nature and hydrocarbon skeleton. Steroids have very small electronegativity values as bond lengths are short and the electrons within are shared (relatively) evenly. As a result, they have a very small dipole moment (charge). Once through the plasma membrane, the steroids can access receptor sites in the cytoplasm or the nucleus.
When a steroid enters a cell and binds to a receptor site, the ensuing conformational change results in an interaction with cell DNA. The outcome is usually an altering of gene expression (specifically transcription and/or translation). Once the DNA of a cell has been infiltrated, a wide array of biological processes can be manipulated (Sever and Glass, 2013).
Prednisolone, a commonly prescribed steroid in cases of chronic obstructive pulmonary disease (COPD) exacerbation, will bind to cytoplasmic receptors and modify cell DNA synthesis. After binding to the glucocorticoid receptors, the inflammatory cells and mediators are inhibited. Following the suppression of these inflammatory processes, respiratory function can improve.
Transporter/carrier molecules
Many bodily functions that are controlled by neurotransmitter release use a method of reuptake to moderate effects. These processes are controlled by enzymes which are sometimes assisted by transporter molecules. Without these actions, the neurotransmitters would retain high concentrations in the synapse and lead to toxicity. An astute method of therapeutic modification involves the inhibition of said enzyme; this allows for a higher concentration of the neurotransmitter to be available to bind to receptor sites and maintain a physiological action. Fluoxetine, a commonly prescribed antidepressant, is a class of selective serotonin reuptake inhibitor (SSRI), which acts by blocking the serotonin transporter (SERT) (Peng et al, 2014). Tricyclics, such as amitriptyline, block the reuptake of noradrenaline by inhibiting the noradrenaline transporter (NAT) (Tatsumi et al, 1997). In the treatment of Parkinsonism, Amantadine maintains a concentration of dopamine by blocking its reuptake (Blanpied et al, 2005).
Antimicrobial pharmacology
The field of antibiotics is not one that can easily be presented here as the different classes all act in very different and specific ways. However, since they are among the most commonly prescribed, beta-lactams are provided as a case study.
All bacteria are prokaryotic, meaning their cells contain no distinct nucleus. Most biologically significant bacteria have a peptidoglycan layer outside their plasma membrane. This layer made of sugars and amino acids provides the target for many antibiotics. If the peptidoglycan layer can be destroyed or damaged, the cell contents will be exposed and perish, meaning the cell can no longer replicate. Within the peptidoglycan layer, there are a number of proteins that link peptides together to create a mesh and envelope the cell. Penicillin comes from a family of drugs known as beta-lactam antibiotics due to the unique beta-lactam ring which forms part of their structure.
Beta-lactam antibiotics selectively bind at the same locations where the peptides expect to form the links; by binding at these key locations, the cell loses its structural integrity and the cell dies. If the concentration of the antibiotic is insufficient to completely eradicate the bacteria, resistance may occur. The most common method in this case is the synthesis of an enzyme (beta-lactamase), which binds with this class of antibiotic and renders them inactive (Lobanovska and Pilla, 2017). The danger of misuse and the potential for resistance was first highlighted by Sir Alexander Fleming in his Nobel prize acceptance speech in 1945.
Conclusion
The principles of pharmacodynamics have been explored from the molecular to cellular level. The interactions of drugs based on chemical structure have also been discussed: whether this is by action at a receptor site on the surface of a cell, by passing through the cell membrane and influencing DNA, inhibiting the molecules which act as transporters, or by simply destroying the wall of a cell and preventing replication. These are the generally accepted means by which most drugs will act and this knowledge allows us to adapt the mechanisms and apply them to other drugs one might come across as a paramedic prescriber.