
Sickle cell disease (SCD) is the name given to a group of inherited conditions affecting the red blood cells (erythrocytes). It causes anaemia, episodes of intense pain, as well as conditions such as acute chest syndrome, splenomegaly, gallstones, sight loss and others (Dormandy et al, 2018). SCD increases the utilisation of health services and causes severe lifestyle problems. This is owing to the complexities of treatment alongside factors such as a decrease in social activities because of exacerbated SCD conditions (Thompson, 2006).
It is estimated that people with SCD will live 22 years less than those without SCD because of the chronic complications associated with the disease, which also translate to a reduction in lifetime income (Lubeck et al, 2019).
These long-term complications affect vital organs causing splenic and renal impairment, increased susceptibility to infections and pulmonary hypertension leading to acute chest syndrome (ACS). All of these contribute to declining health and, in some situations, death (Onimoe and Rotz, 2020).
In addition to reduced life expectancy, the quality of life in a person with SCD is diminished because of long-term morbidities including vaso-occlusive crises causing pain, fatigue and depression. All these can reduce functioning at school and socially (Lubeck et al, 2019).
If the symptoms of a crisis are seen in practice, it is imperative to identify and treat the patient early to alleviate symptoms and address the life-threatening complications discussed in this report.
Case studies play an important role in medicine, teaching students and professionals about important clinical observations concerning rare diseases that are not covered in large-scale studies because of low prevalence rates or ethical concerns (Carey, 2010).
Case studies have come under academic scrutiny and are frequently placed on the lowest rung of study design hierarchies, below studies that find averages in large populations (Carey, 2010). However, analytical and detailed case studies can illuminate fundamental concepts, revealing avenues for development, and broaden and deepen knowledge and understanding of diseases, their processes and treatments (Kanter, 2010).
This case report explores the pathophysiology of SCD, using the comprehensive medical model to assess the patient and draw differential diagnosis and treatment for patients with SCD according to the UK prehospital guidelines.
Case report
A 10-year-old Afro-Caribbean boy arrived home feeling unusually tired after playing football at school. He went straight to sleep but was woken at 17:00 with a sudden onset of intense pain (rated at 10/10) all over his body, primarily in the joints and abdomen. The pain was worse on movement and not relieved by paracetamol.
Most of the patient's history was taken from his adoptive mother, who knew very little about his family history. She confirmed the patient had felt nauseated.
Use of the medical model for comprehensive assessment and history-taking indicated a sickle cell crisis. This was recorded alongside the history, clinical findings, examination, differential diagnosis and appropriate treatments (Box 1) (observations in Table 1).
| Airway | Clear |
| Breathing | Respiratory rate: 24 |
| Circulation | Heart rate: 120 bpm |
| Disability | Glasgow Coma Scale: 15/15 |
| Examination/exposure | Pain on inspiration |
| Paediatric Early Warning Score | 4 |
SPO2=oxygen saturation; bpm=beats per minute
Discussion
The patient in this case report was an Afro-Caribbean boy who raised the index of suspicion for SCD. SCD is an inherited condition mostly affecting people of African or Afro-Caribbean heritage (Thompson, 2006).
The most recent comprehensive public health review of SCD in the UK, which took place in 2016, found 14 000 people with SCD, equivalent to 1 in 4600 people (Dormandy et al, 2018).
Paramedics must ensure they understand key concepts of the disease enabling them to draw upon this in practice; therefore, a thorough understanding of SCD pathophysiology is essential for treatment (Health and Care Professions Council (HCPC), 2018).
Genetics and hereditary aspects
SCD is inherited through an autosomal recessive gene from parents who both carry the gene; therefore, there are far more carriers than people with the condition (NHS, 2019). A child born to parents who both carry the gene has a one in four chance of the genes being expressed and being born with SCD (NHS, 2019). People with one copy of the gene who are said to have the sickle cell trait do not usually experience SCD symptoms.
SCD affects red blood cells (erythrocytes). Haemoglobin is the protein found in erythrocytes; the genes for its amino acid chain consist of a pair of alpha and beta chains located on chromosomes 6 and 11 respectively (Cohn and Roth, 1996).
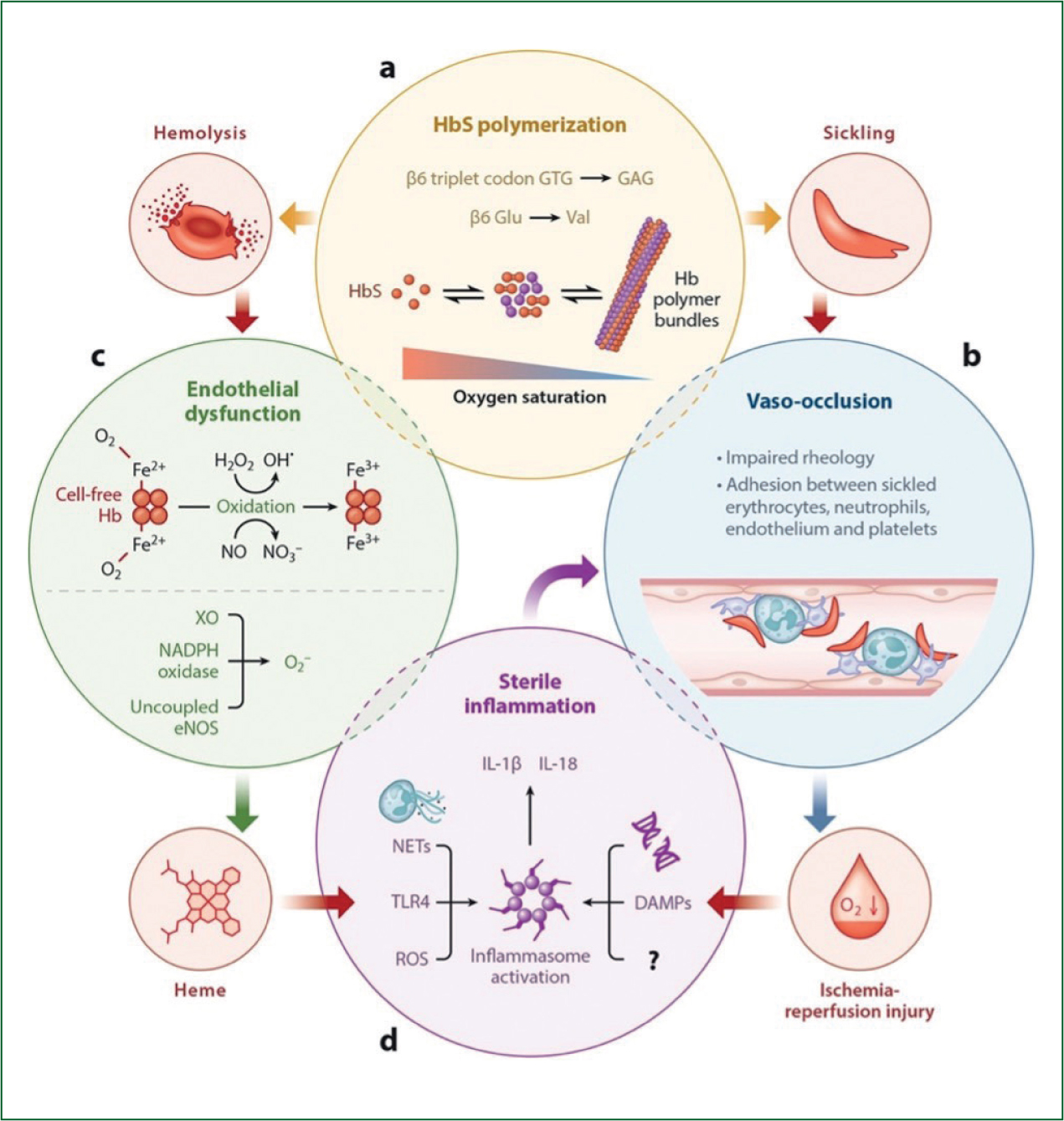
SCD involves a disorder of the beta chain on chromosome 11, which results in the hydrophilic glutamate residue being replaced by a hydrophobic valine residue at the sixth position in the beta chain (Cohn and Roth, 1996). This hydrophobic valine residue causes the haemoglobin to undergo reduced tension when deoxygenated, which changes the structure of erythrocytes to a less soluble, sickle-shaped longer polymer; this is called polymerisation (Thompson, 2006). Polymerisation reduces haemoglobin's binding properties to oxygen, and this reduction in binding is exacerbated when oxygen saturations fall below 85% (Cohn and Roth, 1996).
Malformation of erythrocytes
Polymerised erythrocytes are sickled in shape with adhesive surface antigens that adhere to endothelium cell wall molecules, contributing to a multistep and multicellular cascade causing microvasculature occlusions (Manwani and Frenette, 2013; Maakaron, 2021).
Sickled cells are extremely susceptible to haemolysis, which causes chronic anaemia (Kato et al, 2017). Haemolysis of sickled erythrocytes occurs because polymerised erythrocytes are fragile and this, combined with the trauma of circulation, causes them to rupture more easily (Braunstein, 2020). As polymerised erythrocytes are more fragile than non-polymerised erythrocytes, they have a lifespan of 16 days rather than 120 days, thereby reducing the total number of erythrocytes, leading to anaemia.
As a result of chronic anaemia, bone marrow undergoes stress to produce erythrocytes. This results in the production of immature erythrocytes known as reticulocytes, which have molecular adhesion factors (Sundd et al, 2019). In a condition classed as uncompensated haemolytic anaemia, the bone marrow can no longer compensate with the correct type and number of erythrocytes (Braunstein, 2020).
The increase in demand on the spleen to filter the haemolysed erythrocytes, along with the production of reticulocytes with adhesive factors, contribute to vaso-occlusion causing splenomegaly (Corwin, 2008; Braun and Anderson, 2017). Vaso-occlusion in the spleen blocks the flow of blood within it, causing painful tissue ischaemia.
There is a reduction in splenic immune function but an increase in size because of the accumulation of haemolysing erythrocytes in the microvasculature over time (Conran and Belcher, 2018; Braunstein, 2020).
Endothelial dysfunction
Complications involving the endothelium and erythrocytes in SCD lead to multiple cardiovascular system problems (Kato et al, 2017). The endothelium is the inner lining of the blood vessels and is in direct contact with circulating cells. Through the release of relaxing and contracting factors such as nitric oxide, it plays a major role in blood fluidity, platelet aggregation and vascular tone (Félétou, 2011).
Haemolysis of erythrocytes damages the endothelium directly while chronic anaemia affects the cardiovascular system (Teixeira et al, 2017). Nitric oxide is critical for maintaining vascular tone; however, in SCD, the bioavailability of nitric oxide is severely impaired as a direct result of haemolysis (Teixeira et al, 2017).
During haemolysis, erythrocytes release free haemoglobin, which scavenges nitric oxide and causes excessive production of reactive oxygen species (ROS). ROS oxidises nitric oxide, further depleting stores and altering the ability of the endothelial to function (Teixeira et al, 2017).
Reperfusion injury of the endothelium occurs when blood flow is restored to tissues following vaso-occlusion. Reoxygenating the tissues causes a release of ROS, inducing an inflammatory response of white blood cells and platelets (Conran and Belcher, 2018). These adhere to sickled red blood cells, aggregate on endothelial cells within blood vessels and contribute to endothelium dysfunction (Conran and Belcher, 2018).
Adhesion of stress reticulocytes from expressed surface proteins
Impaired rheology from cellular adhesion of erythrocytes, neutrophils and endothelial cells plays an important role in the initiation of vaso-occlusion (Figure 1) (Manwani and Frenette, 2013).
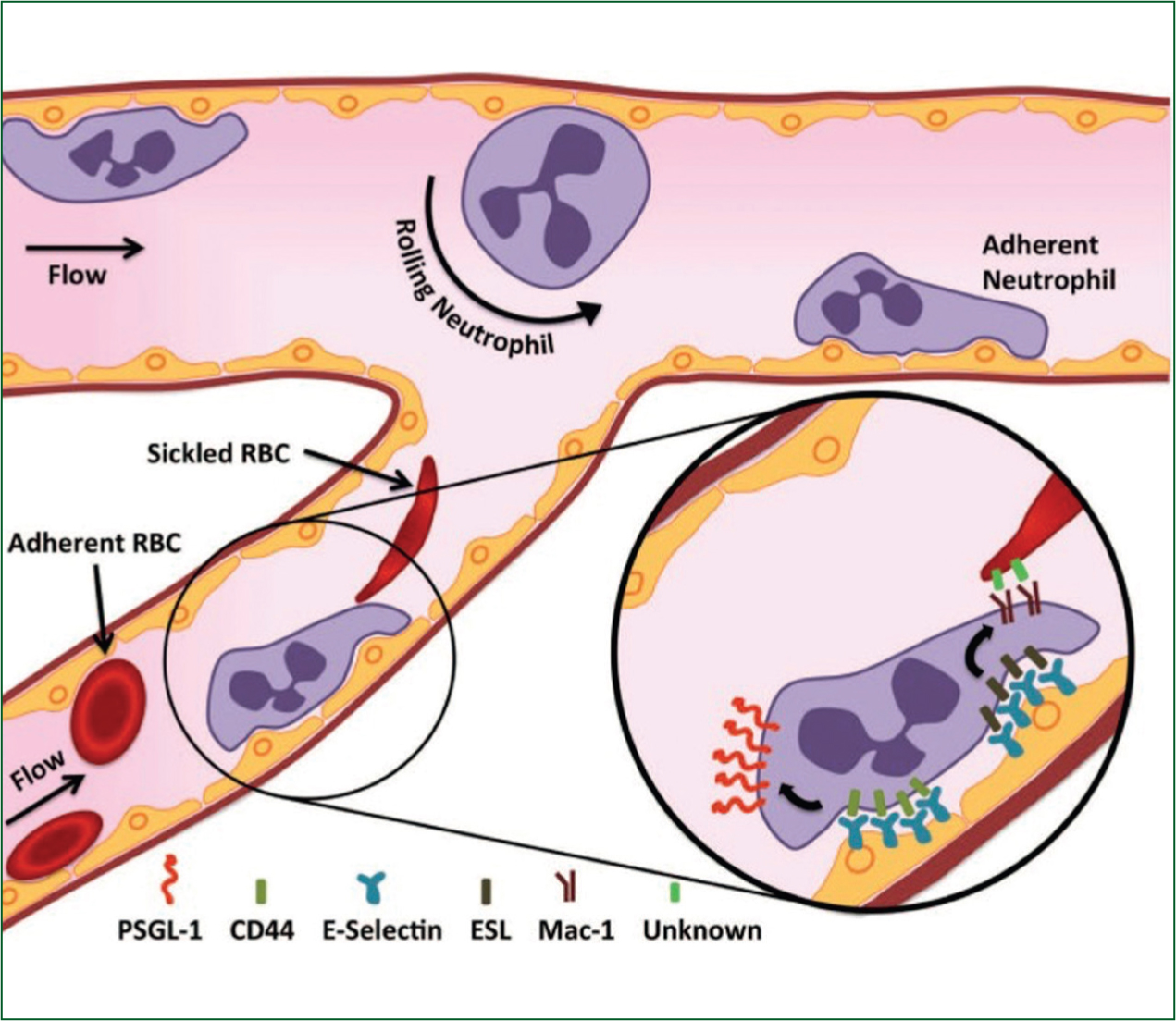
Studies have highlighted how sterile inflammation follows tissue ischaemia from vaso-occlusion. The release of damage-associated molecules following oxidation of haemoglobin and the adhesion of sickled erythrocytes activates potent inflammation reactions, increasing coagulopathy (Conran and Belcher, 2018). These inflammatory reactions are known as sterile inflammation (Sundd et al, 2019).
The process is highly dynamic with multiple contributory physiological factors, with evidence suggesting that the process can be precipitated by emotional stress, cold weather or physical exertion (Manwani and Frenette, 2013). The combination of sickled erythrocytes, neutrophils released during inflammation and reticulocytes contribute to an increase in plasma viscosity. The adhesiveness of endothelium and the increase of leucocytes combine to form vaso-occlusions in microvasculature resulting in painful tissue hypoxia and ischaemia (Sundd et al, 2019).
Assessing and diagnosing patients with SCD is important to ensure they are started on the correct treatment path. This relies on comprehensive history-taking and thorough physical assessments to understand the symptoms. From the history gained in this case report, it is known that the child is of Afro-Caribbean ethnicity and experiencing sudden, severe pain all over his body while denying any trauma. The pain is worse in his joints and abdomen.
The existence of joint pain alone is a non-specific symptom for a variety of conditions but performing a thorough abdominal assessment revealed upper left quadrant pain and splenomegaly. The existence of pain in the joints and splenomegaly in the patient is suggestive of vaso-occlusion in the microvasculature, causing recurrent and unpredictable episodes of tissue ischaemia in the spleen and in various joints resulting in intense pain and vascular necrosis (Okpala and Tawil, 2002).
The patient's history, alongside the knowledge that SCD primarily affects people of African or Afro-Caribbean heritage, raises the index of suspicion that the patient is having a sickle cell crisis (Thompson, 2006).
There are several complications associated with SCD, primarily ACS. ACS occurs when the sickled erythrocytes adhere to the pulmonary microvasculature and cause occlusions and tissue hypoxia (Jain et al, 2017). Tissue hypoxia induces the inflammatory response, causing congestion of the cardiopulmonary system owing to vaso-occlusions in the arterioles. This reduces the amount of gaseous exchange that can take place, while causing acute inflammation of the airways because of the proximity of the vasculature (Jain et al, 2017).
The presentation of ACS includes tachypnoea, tachycardia, pyrexia and hypoxia with basal crackles in the lungs on auscultation (Brown et al, 2019). During the assessment of this patient, all these physiological indicators were present as well as increased pain on inspiration.
ACS is the leading cause of death in patients with SCD, so it is imperative that they are identified and action taken (Brown et al, 2019).
Although no clinical signs of anaemia were identified in the boy in this case study, the history (Box 1) indicated the patient was susceptible to infections, which are common in patients with SCD because to reduced splenic function associated with sickle cell-induced anaemia (Conran and Belcher, 2018). SCD is commonly tested for in pregnancy and at birth within the NHS. However, the patient in question was adopted and his adopted mother did not know whether SCD occurred in his biological relatives, nor were there any details confirming a diagnosis of SCD or other medical conditions.
Forming a differential diagnosis allows clinicians to reduce interpretation errors as well as understand how a single diagnosis may cover multiple phenotypes (Cook and Décary, 2020). The knowledge that a disease or condition may involve multiple phenotypes is evident in this case, with the patient experiencing severe pain and respiratory concerns. While a sickle cell crisis causes intense pain in the joints, similar pain is present in conditions such as osteomyelitis and septic arthritis, making diagnosis difficult without reviewing all the available evidence (BMJ Best Practice, 2022).
The fact that SCD patients are at a higher risk of osteonecrosis can further compound the difficulties in identifying an individual in crisis (Kato et al, 2017). Several physiological indicators were present when this patient was assessed, including evidence of symptoms associated with pulmonary embolisms and respiratory infections (Maakaron and Taher, 2020). It is important to be able to identify these as phenotypes present when an individual is in crisis, not as a separate diagnosis.
Diagnosis, a necessary component of clinical decision-making, involves identifying the aetiology of a disease or condition through evaluating physiological indicators explored through history-taking and physical examination (Cook and Décary, 2020). Understanding the aetiology of SCD and applying this knowledge regarding the multiple phenotypes of an individual in crisis ensures the right diagnosis is made; this is vital in ensuring appropriate treatment is initiated. This knowledge, alongside information gained from history-taking, identification of physiological indicators and findings from patient examination, allowed SCD to become the working diagnosis.
There are several treatments for SCD, with therapies focusing on the polymerisation of abnormal erythrocytes through regular transfusions and targeted drug therapy; erythrocytes are significant in the cascade of conditions they can cause (Manwani and Frenette, 2013). The main aim is to reduce the number and severity of vaso-occlusion events. These events can have detrimental effects leading to an inflammatory response and spleen problems, increasing susceptibility to sepsis and further complications (Conran and Belcher, 2018).
Current pharmacological guidelines emphasise the use of opioids, non-steroidal anti-inflammatory drugs (NSAIDs) and paracetamol for acute pain during a sickle cell crisis. The American Society of Hematology (ASH) recommends several non-pharmacological interventions (Brandow et al, 2020), including patient coping mechanisms such as relaxation therapies, massage, yoga, cognitive behavioural therapy, meditation and the use of virtual reality, alongside pharmacological management. It has also been noted that non-pharmacological therapies may be tailored to paediatric patients (Brandow et al, 2020).
Although long-term therapies have been shown to improve quality of life, they also limit an individual's social activities as regular hospital appointments are essential (Thompson, 2006; Howard, 2016).
This report concerns the prehospital environment so focuses on the treatments available to paramedics in the UK. The Joint Royal Colleges Ambulance Liaison Committee (JRCALC) guidelines emphasise the use of patient guidance. This includes paying attention to a patients' personalised treatment plan; patients are likely to know which medications work best for them and will often have their targeted analgesia available (Brown et al, 2019).
Prehospital clinicians must adhere to guidelines and treat symptoms within their scope of practice, most noticeably providing pain management (Brown et al, 2019). Patients who are known to have SCD may have their own treatment plan for a crisis. This should be followed as it is tailored to the individual with appropriate and effective analgesia (BMJ Best Practice, 2022).
In this case, the problems identified during the patient's assessment consisted of low oxygen saturations, tachypnoea, basal crackles, reduced peripheral capillary refill time, pyrexia and 10/10 pain not relieved by paracetamol. A systematic, rigorous primary survey should be used to assess and treat the patient alongside clinical guidelines (HCPC, 2018).
Breathing needs to be corrected with high-flow supplementary oxygen to counter tissue hypoxia and ACS as it is safer to over-oxygenate until a reliable oxygen saturation (SPO2) measurement is available (Brown et al, 2019). However, recent studies have cautioned against prolonged nitrous oxide (Entonox) use (Desprairies et al, 2020) because of its known effect on vitamin B12 metabolism, leading to severe neurological neuropathy and vitamin B12 deficiencies. It is appropriate to administer Entonox while preparing more suitable opioid analgesia in this clinical situation because there is no history of reported or continuous use of Entonox, which minimises the risk of vitamin B12 deficiency (Desprairies et al, 2020).
In the context of severe pain, the use of strong analgesia such as opioids via subcutaneous or oral administration is recommended in addition to NSAIDs and paracetamol (National Institute for Health and Care Excellence, 2012; Brown et al, 2019).
Although subcutaneous administration reduces the bioavailability of the analgesia, SCD patients are more susceptible to infections. While paramedics are now able to provide aseptic intraveous cannulation, a less invasive procedure with the same or similar analgesic effect is favourable for paediatric patients (Braunstein, 2020). Administration of 3 mg morphine sulphate in a 0.30 ml subcutaneous solution is appropriate, with a once-only repeat dose of 3 mg in 0.30 ml after 60 minutes of the first dose. It is recommended that practitioners consider ondansetron for opioid-induced nausea and vomiting is recommended (Brown et al, 2019).
Intravenous sodium chloride 0.9% has historically been used as analgesia as its actions promote cellular hydration and decrease the blood viscosity, slowing the sickling process of erythrocytes (Okomo and Meremikwu, 2017). However, there are no high-quality studies to support this, and JRCALC guidelines indicate fluid resuscitation only if there are clinical signs of dehydration. Fluid therapy was not used in this case as observations (Figure 2 did not indicate dehydration (Okomo and Meremikwu, 2017; Brown et al, 2019).
The ASH does not recommend the use of fluids in acute sickle cell crisis but does note the potential risks of administering them to patients with decreased cardiopulmonary function, which potentially increases pulmonary oedema (Brandow et al, 2020). Decreased cardiopulmonary function may detrimentally affect patients with clinical signs of ACS; the risk is greater in adults than in children (Brandow et al, 2020).
Conclusion
SCD is a serious, hereditary disease consisting of multiple complex conditions. Early recognition through clinical investigation is vital in reducing the cascade of events that cause morbidity and mortality (Manwani and Frenette, 2013; Braunstein, 2020).
Understanding the pathology of disease ensures paramedics can use their knowledge to identify sickle cell crises. Skills inform practice; initiating the correct treatment is essential in reducing potentially fatal complications that may arise if SCD remains untreated (HCPC, 2018; Sundd et al, 2019).